2.11. Automated Convergence
| download: | pdf |
|---|
The accuracy of computed materials properties such as equilibrium lattice parameters, binding energies, and elastic moduli depends on a variety of computational parameters, most notably the quality of plane wave basis sets and the density of k-meshes for integrations in reciprocal space.
The optimal choice of these parameters depends both on the material under investigation and on the properties of interest. For example, metallic aluminum requires very fine k-meshes to achieve convergence of the total energy while semiconductors are well described by a rather coarse k-mesh. Finding the most appropriate parameter setting is important, but tedious to do by hand. The Automated Convergence Module automates the process of determining optimal parameter settings in Vasp for achieving the desired level of accuracy in calculations of materials properties.
Activate the Convergence menu entry in MedeA by clicking Tools >> Automated Convergence. Next select Convergence >> VASP computation to start a Convergence job or Convergence >> Monitor to analyze results.
2.11.1. Submitting VASP Convergence Jobs
The Convergence run interface looks like this:
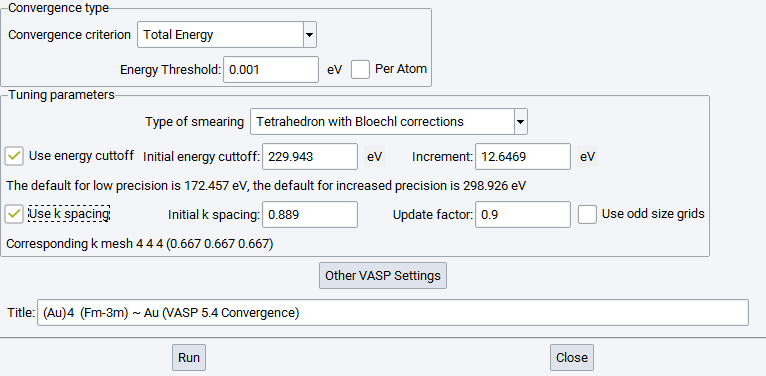
Convergence type
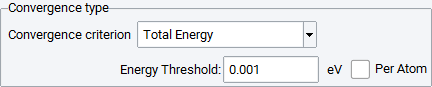
You can use the following criteria with Convergence:
Total Energy: Converges the VASP total energy, where you can select a value as convergence threshold, either for the whole system or per atom.
Structure Optimization: Converges both the single-point stress and the lattice parameters and atomic positions. You can select to Relax atoms only.
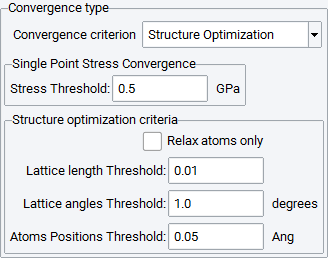
The convergence will be studied as follows:
- Single point stress convergence calculations with the stress tensor (Stress Threshold in GPa) as the convergence criterion
Using results from a Structure optimization with convergence based on the relative change of the cell length, angles and atomic positions. To keep the cell fixed, select Relax atoms only
2.11.2. Tuning Parameters
Using the above convergence criteria the module will optimize the following parameters:
Energy cutoff (Plane wave cutoff, PWC): Starting from the initial energy cutoff the module will increase the cutoff by Increment until convergence is reached. VASP potentials provide a range of cutoff energies for each atom, defined by the limits EMIN and EMAX. The default value is the average between EMIN and EMAX. For systems with more than one type of atoms, the average between the highest EMIN and the highest EMAX is taken. The default increment is 1/10 of the difference between these two values.
K spacing (k-point spacing, KPS): The initial value of the KPS is set by the parameter k-spacing in such a way that the resulting k-mesh is at least 3x3x3 to allow using the tetrahedron method. In the refinement of the k-mesh, the initial k-spacing is multiplied by the Update factor until the actual number in the k-mesh increases.
Smearing width: You can optimize this parameter only when Methfessel-Paxton k-integration is chosen. The default is to use the tetrahedron method with Bl\({\ddot{o}}\) chl corrections, which has no smearing parameter. The update factor decreases the smearing width during the convergence process.
2.11.3. Monitoring VASP Convergence Jobs
Choosing a job
The Monitor menu queries the selected JobServer for a list of convergence jobs. Select a Job to retrieve and display the results from an ongoing or finished job.
Multi-parameter convergence
Before detailing the content of the monitor window, let us explain the progress of a convergence Job.
As long as only one parameter is used for tuning, the convergence process simply consists in updating the parameter until convergence is reached. If, however, several parameters are used simultaneously, the parameter space is multi-dimensioned and there might be several paths that lead to different points, where convergence is reached.
The strategy used for our problem is intended to limit the overall cost as follows. For the sake of simplicity, let us consider 2 tuning parameters p1 and p2, but this might be extended directly to any number of parameters. Two series of independent tasks are started with the following settings: in the first series, p2 is set to its initial value and p1 is tuned until convergence is reached after n1 steps; in the second series, p2is converged in n2 steps while is p1 is unchanged. Finally two tasks are launched with the two last values of both parameters: (p1 (n1-1), p2 (n2-1)) and (p1 (n1), p2 (n2)) and the convergence is tested again with the results of these two tasks. This strategy allows running at least as many tasks in parallel as there are tuning parameters, and even more during the early stage of the job.
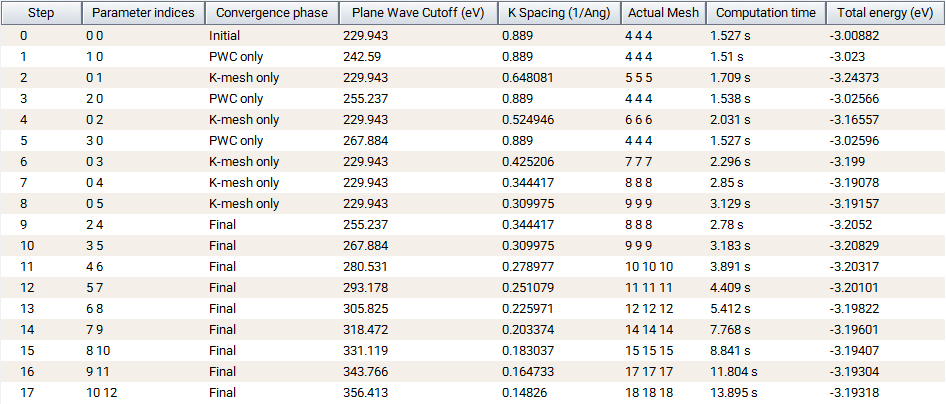
Monitor window.
This window presents a report table and a set of graphics. The table contains a line for each step (VASP task), displaying the parameter values and the computed properties.
The graphics frame contains a graph for each single-parameter convergence axis and one for the final convergence steps (with all parameters):
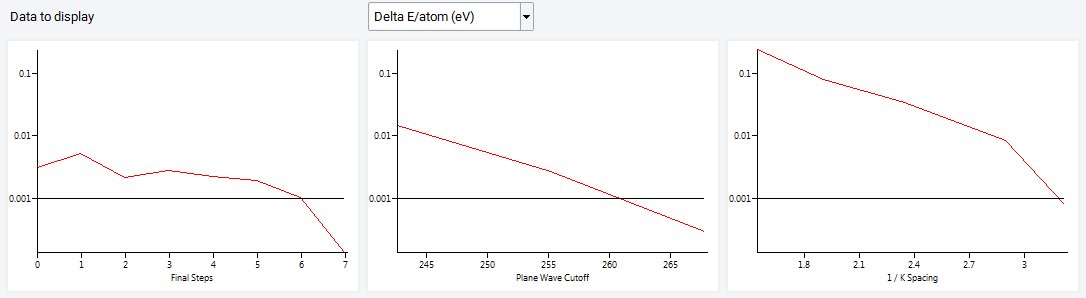
The selection of columns can be adjusted via Column Display: Parameter indices, Convergence phase, K Spacing, Actual K Spacing, Actual mesh, Computation time, and Total energy and differences per atom and cell.
When the monitored job is running, some of the computed values are still missing and are indicated by a - symbol.
An estimate of the minimum completion time is calculated by multiplying the time of the longest completed task by the minimum number of remaining tasks.
Basic features of the Automatic Convergence module are also found in Point Defect Analysis tool.
| download: | pdf |
|---|