3.1. Density Functional Theory
Contents
| download: | pdf |
|---|
3.1.1. The Kohn-Sham Equations
In the mid-1960’s, Hohenberg & Kohn [1] and Kohn & Sham [2] formulated a rather remarkable theorem, which states that the total energy of a system such as a solid, a surface, and a molecule depends only on the electron density of its ground state. In other words, one can express the total energy of an atomistic system as a functional of its electron density
The idea of using the electron density as the fundamental entity of a quantum mechanical theory of matter originates in the early days of quantum mechanics in the 1920’s, especially by the work of Thomas [3] and Fermi [4]. However, in the subsequent decades, it was rather the Hartree-Fock approach [5], which was developed and applied to small molecular systems. Calculations on realistic solid state systems were then out of reach. In 1951, Slater [6] used ideas from the electron gas with the intention to simplify Hartree-Fock theory to a point where electronic structure calculations on solids became feasible. Slater’s work, which led to the so-called \({X_{\alpha}}\)-method [7] has contributed tremendously to the development of electronic structure calculations.
However, the ad hoc character of the \({X_{\alpha}}\)-approach, combined with other simplifications in the so-called scattered-wave method have fueled heated disputes about the “ab initio” character of this approach, which was carried over into the comparison of density functional theory vs. Hartree-Fock based methods. Today’s density functional methods can be considered “ab initio”. In fact, recent methods such as LDA+U for correlated systems and hybrid functionals and screened-exchange for excitations in semiconductors synthesize ideas of both theoretical approaches, the wavefunction based Hartree-Fock concept and density functional theory.
In solid-state systems, molecules, and atoms, the electron density is a scalar function defined at each point r in real space,
The electron density and the total energy depend on the type and arrangements of the atomic nuclei. Therefore, one can write
The set (\(R_{\alpha}\)) denotes the positions of all atoms, \(\alpha\) in the system under consideration. Eq is the key to the atomic-scale understanding of electronic, structural, and dynamic properties of matter. If one has a way of evaluating expression one can, for example, predict the equilibrium structure of solid, one can predict the reconstruction of surfaces, and the equilibrium geometry of molecules adsorbed on surfaces.
Furthermore, the derivative of the total energy Eq. (3) with respect to the nuclear position of an atom gives the force acting on that atom. This enables the efficient search for stable structures and, perhaps more importantly, the study of dynamical processes such as diffusion or the reaction of molecules on surfaces. Most of the considerations discussed here are based on the Born-Oppenheimer approximation in which it is assumed that the motions of the electrons are infinitely faster than those of the nuclei. In practice, this means that the electronic structure is calculated for a fixed atomic arrangement and the atoms are then moved according to classical mechanics. This is a fairly good approximation for heavy atoms like W, but may cause errors for light atoms such as H or Li.
In density functional theory, the total energy is decomposed into three parts, a kinetic energy, an electrostatic or Coulomb energy, and a so-called exchange-correlation energy,
The most straightforward term is the Coulomb energy U. It is purely classical and contains the electrostatic energy arising from the Coulomb attraction between electrons and nuclei, the repulsion between all electronic charges, and the repulsion between nuclei
With
where e is the elementary charge of a proton and \(Z_{\alpha}\) is the atomic number of atom \(\alpha\). The summations extend over all atoms and the integrations over all space. Once the electron density and the atomic numbers and positions of all atoms are known, expression - can be evaluated by using the techniques of classical electrostatics.
The kinetic energy term, \(T_{0}\), is more subtle. In density functional theory, the “real” electrons of a system are replaced by “effective” electrons with the same charge, mass, and density distribution. However, effective electrons move as independent particles in an effective potential, whereas the motion of a “real” electron is correlated with those of all other electrons. \(T_{0}\) is the sum of the kinetic energies of all effective electrons moving as independent particles. Often, one does not explicitly make this distinction between real and effective electrons.
If each effective electron is described by a single particle wave function, \({\Psi_{i}}\), then the kinetic energy of all effective electrons in the system is given by
Expression the sum of the expectation values of one-particle kinetic energies; \(n_{i}\) denotes the number of electrons in state i. By construction, dynamical correlations between the electrons are excluded from \(T_{0}\).
The third term of eq, called exchange-correlation energy, \(E_{xc}\), includes all remaining complicated electronic contributions to the total energy. The most important of these contributions is the exchange term. Electrons are Fermions and obey the Pauli exclusion principle. In real space, the Pauli principle implies that around each electron with a given spin, all other electrons with the same spin tend to avoid that electron. As a consequence, the average Coulomb repulsion acting on that electron is reduced. This energy gain is called exchange energy. Effectively each electron is surrounded by a positive exchange hole. Slater showed that the total charge integrated over the entire exchange hole equals is \(+e^{162}\). By definition, the additional many-body interaction terms between electrons of opposite spin are called correlation energy.
As an illustration, the total energy of a single C atom is approximately -1019 eV, that of a Si atom -7859 eV and that of a W atom -439634 eV. The kinetic energy and the Coulomb energy terms are of similar magnitude but of opposite sign whereas the exchange-correlation term is about 10% of the Coulomb term and attractive for electrons (because the exchange-hole is positive). The correlation energy is smaller than the exchange energy but plays an important role in determining the details in the length and strength of interatomic bonds. In fact, compared with the total energy, the binding energy of an atom in a solid or on a surface is quite small and lies in the range of about 1 to 8 eV. Energies involved in changes of the position of atoms, such as in the reconstruction of a surface can be even smaller. For example, only about 0.03 eV are required to flip an asymmetric Si-dimer on a reconstructed Si(001) surface from one conformation into another where the role of the upper and lower Si atoms is reversed. It is a tremendous challenge for any theory to cope with such a range of energies. Density functional theory, as it turns out, comes amazingly close to this goal.
The Hohenberg-Kohn-Sham theorem, which is a central part of density functional theory, states that the total energy is at its minimum value for the ground state density and that the total energy is stationary with respect to first-order variations in the density, i.e.
In conjunction with the kinetic energy, we have introduced one-particle wave functions \(\Psi_{i}\), which generate the electron density
As in the expression for the kinetic energy, ni denotes the occupation number of the eigenstate i, which is represented by the one-particle wave function \(\Psi_{i}\). So far, one has a formally exact theory in the sense that no approximations have been made yet to the many-electron interactions. By construction, \({\rho}\) in Eq. (11) is the exact many-electron density.
The goal of the next step is the derivation of equations that can be used for practical density functional calculations. Through these equations and we have introduced one-particle wave functions. A change of these wave functions corresponds to a variation in the electron density. Therefore, the variational condition (10) can be used to derive the conditions for the one-particle wave functions that lead to the ground state density. To this end, one substitutes Eq. (11) in expression (10) and varies the total energy with respect to each wave function. This procedure leads to the following equations:
with
Equations are called the Kohn-Sham equations. The electron density, which corresponds to these wave functions, is the ground state (12) density, which minimizes the total energy. The solutions of the Kohn-Sham equations form an orthonormal set, i.e.
This additional constraint is achieved by introducing Lagrange multipliers, \(\epsilon_{i}\), in Eq. (12). These “Lagrange multipliers” are effective one-electron eigenvalues and their interpretation will be discussed later. These eigenvalues are used to determine the occupation numbers \(n_{i}\) by applying the Aufbau principle. The eigenstates are ordered according to increasing eigenvalues. For non-spin polarized systems each state is occupied by two electrons until all electrons are accommodated. In spin polarized systems, each state is occupied by at most one electron.
As a consequence of the partitioning of the total energy (14), the Hamiltonian operator in the Kohn-Sham equations (12) contains three terms, one for the kinetic energy, the second for the Coulomb potential, and the third for the exchange-correlation potential.
The kinetic energy term is the standard second-order differential operator of one-particle Schödinger equations and its construction does not require specific knowledge of a system. In contrast, the Coulomb potential operator, \(V_{c}\), and the exchange-correlation potential operator, \(\mu_{xc}\), depend on the specific electron distribution in the system under consideration.
The Coulomb or electrostatic potential \(V_{c}\) at point \(r\) is generated from the electric charges of all nuclei and electrons in the system. It can be evaluated directly in real space,
In condensed systems it is more convenient to use Poisson’s equation
to calculate the electrostatic potential. Here, \(q\) denotes both the electronic charge distribution \({\rho}\) and the positive point charges of the nuclei at positions \(R_{\alpha}\).
The exchange-correlation potential is related to the exchange-correlation energy by
Equation (17) is formally exact in the sense that it does not contain any approximations to the complete many-body interactions. In practice however, the exchange-correlation energy (and thus the exchange-correlation potential) is not known and one has to make approximations.
As a consequence of the Kohn-Sham theorem, the exchange-correlation energy depends only on the electron density. As a simple and, as it turns out, surprisingly good approximation one can assume that the exchange-correlation energy depends only on the local electron density around each volume element \(dr\). This is called the local density approximation (LDA)
Below is an illustration of the basic idea. In any atomic arrangement such as a crystal, a surface, or a molecule, there is a certain electron density \({\rho}\) at each point \(r\) in space. The LDA then rests on two basic assumptions:
- the exchange and correlation effects come predominantly from the immediate vicinity of a point \(r\) and
- these exchange and correlation effects do not depend strongly on the variations of the electron density in the vicinity of \(r\).
If conditions (i) and (ii) are reasonably well fulfilled, then the contribution from volume element \(dr\) would be the same as if this volume element were surrounded by a constant electron density of the same value as within \(dr\). This is an excellent approximation for metallic systems, but represents quite a significant simplification in systems with strongly varying electron densities.

Illustration of the local density approximation: For the purpose of computing the exchange-correlation energy in a volume element \(dr_{i}\), the electron density \(\rho_{i}\) around point \(r_{i}\) is assumed to be constant in the near surrounding. Note that the values of \(\rho_{i}\) is different in each volume element.
A system of interacting electrons with a constant density is called a homogeneous electron gas. Substantial theoretical efforts have been made to understand and characterize such an idealized system. In particular, the exchange-correlation energy per electron of a homogeneous electron gas, \(\epsilon_{xc}^{0}(\rho)\) has been calculated by several approaches such as many-body perturbation theory by Hedin and Lundqvist [8], quantum Monte-Carlo methods by Ceperley and Alder [9]. As a result, \(\epsilon_{xc}(\rho)\), is quite accurately known for a range of densities. For practical calculations, \(\epsilon_{xc}(\rho)\) is expressed as an analytical function of the electron density. There are various analytical forms with different coefficients in their representation of the exchange-correlation terms. These coefficients are not adjustable parameters, but rather they are determined through first-principles theory. Hence, the LDA is a first-principles approach in the sense that the quantum mechanical problem is solved without any adjustable, arbitrary, or system depended parameters.
Explicit forms for the local density exchange, originally given by Gàspàr [10] and Kohn & Sham [11]. Correlation terms are according to Hedin & Lundqvist [12]. Exchange and correlation energies per electron are denoted by \({\epsilon}\) and the corresponding potentials by \({\mu}\). Both quantities are given in Hartree atomic units (1 Hartree = 2 Rydberg = 27.21165 eV). The units for the electron density are number of electrons/(Bohrradius)3.
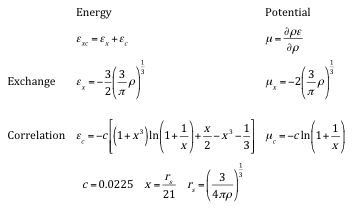
Note that there are two types of exchange-correlation terms, one for the energy and one for the potential. The energy, \(\epsilon_{xc}(\rho)\), is needed to evaluate the total energy and the potential term, \(\mu_{xc}(\rho)\), is required for the Kohn-Sham equations. The two terms are, following (17) and (18), related.
Using the explicit formulas given in the table above, one can evaluate the exchange-correlation potential for any electron density \(\rho\). Thus, all terms of the effective one-particle operator in the Kohn-Sham equations are defined and one can proceed with a computational implementation.
3.1.2. Interpretation of One-Particle Energies
The fundamental quantities in density functional theory are the electron density and the corresponding total energy, but not the one-particle eigenvalues. However, the one-electron picture is so useful that one seeks to exploit the Kohn-Sham eigenvalues and one-particle wave functions as much as possible. The one-particle energies of effective electrons have been introduced in the derivation of the Kohn-Sham equations as Lagrange multipliers. The Kohn-Sham equations have the form of an eigenvalue problem in which each wave function has an associated eigenvalue \(\epsilon_{i}\) with an occupation number of \(n_{i}\). Janak’s theorem [13] provides a relationship between the total energy and these eigenvalues.
The eigenvalue \(\epsilon_{i}\) equals the change of the total energy with the change of the occupation number of level i. However, it is desirable to seek a more direct physical interpretation of these eigenvalues. Already before the formulation of present density functional theory, the one-electron picture has become widely used in solid-state and molecular physics. For example, the distinction between a metal and an insulator is based on the analysis of the energy bands (energy bands are one-electron energies plotted as a function of different directions in reciprocal space); the characteristics of semiconductors and semiconductor/metal junctions are explained in terms of energy band structures; photoemission experiments are conveniently interpreted by a one-electron picture, often with quite reasonable quantitative agreement between theory and experiment. Furthermore, the analysis of the s, p, and d character of partial densities of states has become an extremely useful tool in the understanding of chemical bonding in alloys and compounds. Electronic band structures and partial densities of states are easily obtained from MedeA VASP.
Furthermore, at least qualitatively reasonable optical spectra can be obtained making use of the computed Kohn-Sham energy levels, evaluating transitions from occupied to unoccupied states. Here all possible transitions are considered where the wave vectors (or crystal momentum) for the initial and final states are equal, i.e. the direct transitions. For each of these transitions absorption can occur for photons whose frequency corresponds to the energy difference between the occupied and unoccupied states. Integration over all wave vectors gives the joint density of states. The intensity of each transition is then computed from the wave functions for each pair of occupied and unoccupied states for all k-vectors. These matrix elements modify the joint density of states yielding the imaginary part of the dielectric function \(\epsilon_{2}\). From \(\epsilon_{2}\) one obtains the real part of the dielectric function \(\epsilon_{1}\) from the so-called Kramers-Kronig transformation. Knowledge of \(\epsilon_{1}\) and \(\epsilon_{2}\), i.e. the complex dielectric function, is sufficient to derive the absorption coefficient, reflectivity, and the refractive index. In MedeA VASP, the matrix elements are directly computed by VASP. The Kramers-Kronig transformation is performed by VASP providing the complex dielectric function as a function of energy. From this primary information the other optical functions are computed and displayed in MedeA. The dielectric function as computed by VASP is accessible in the OUTCAR file. If desired, the individual transition matrix elements can be accessed there as well.
While the direct interpretation of the Kohn-Sham eigenvalues as excitation energies often gives quantitative agreement with experimental photoemission spectra and reasonable agreement with optical spectra (if excitonic effects are minor), there are significant differences in quantities such as energy band gaps in semiconductors. In fact, discrepancies of over a factor of two can be found between measured values and the LDA eigenvalues. Such discrepancies between experimental excitation energies and differences between LDA eigenvalues are not necessarily a failure of the LDA, but rather an inappropriate interpretation of theoretical results. In the derivation of the Kohn-Sham equations given above, the effective one-particle eigenvalues were never required to be excitation energies! Only the total electron density and the corresponding total energy have rigorous meaning. It is possible, though, to use the results of density functional calculations as input into rigorous evaluations of excitation energies, as shown, for example, by Hybertsen & Louie [14].
The highest occupied electronic level in a metallic system is called the Fermi energy or Fermi level, \(E_{F}\). The nature of the electronic states at \(E_{F}\) play a crucial role in determining materials properties such as electrical and thermal conductivity, magnetism, and superconductivity. The MedeA Electronics module enables visualization of the Fermi surface and provides access to the transport properties. On surfaces, the energy difference between \(E_{F}\) and the electrostatic potential in the vacuum region, \(V_{0}\), above the surface is the work function, \({\phi}\). While in general the Kohn-Sham eigenvalues are not excitation energies, it was shown by Schulte [15] that for a metallic system the highest occupied Kohn-Sham eigenvalue can be directly interpreted as the work function. Thus, the agreement between experimental and calculated work functions provides a good test for the quality of actual calculations. With present LDA approaches, the calculated values are typically within 0.1-0.2 eV of the experimental results.
3.1.3. Spin-Polarization
So far, the discussion of density functional theory was restricted to non-spin-polarized cases. However, many systems such as magnetic transition metals or molecules such as O2 involve unpaired electrons or molecular radicals and thus require a spin-polarized method. In such systems, the number of electrons with “spin-up” can be different from that with “spin-down”. In the early 1970’s, von Barth & Hedin [16] and Gunnarson, Lundqvist & Lundqvist [17] generalized density functional theory to accommodate spin-polarized systems. This resulted in spin density functional theory with the local spin density (LSD) approximation.
Explicit form of local spin density exchange-correlation terms as given by von Barth & Hedin
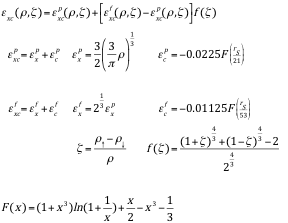
Exchange-correlation potential
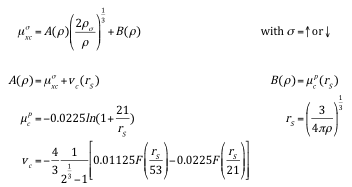 |
| Energies and potentials are given in Hartree atomic units; the units for the electron and spin densities are. |
In the local spin density functional (LSDF) theory, the fundamental quantities are both the electron density, \(\rho\), and the spin density, \(\sigma\). The spin density is defined as the difference between the density of the spin-up electrons and the density of the spin-down electrons
with the total electron density
In LSDF theory, the exchange-correlation potential for spin-up electrons is in general different from that for spin-down electrons. Consequently, the effective potential (13) becomes dependent on the spin. Thus, the Kohn-Sham equations (12) in their spin-polarized form are
\(\sigma = \uparrow\) or \(\downarrow\)
with
The exchange-correlation potential in LSDF theory depends on both the electron density and the spin density, as written in equation (24). There are two sets of single-particle wave functions, one for spin-up electrons and one for spin-down electrons, each with their corresponding one-electron eigenvalues. For the case of equal spin-up and spin-down densities, the spin density is zero throughout space and LSDF theory becomes identical with the LDF approach. Notice that in spin-polarized calculations, the occupation of single-particle states is 1 or 0, but there is still only one Fermi energy. In magnetic systems, the spin-up and spin-down electrons are often referred to as “majority” and “minority” spin systems. The table in the next section gives an example of the local spin density exchange-correlation formula by von Barth & Hedin [18].
3.1.4. Generalized Gradient Approximation
A large number of total energy calculations have shown that the LDA gives interatomic bond lengths within \(\pm 0.05\) of experiment or better for a great variety of solids, surfaces, and molecules. However, the following systematic trends are found:
- most lattice parameters predicted with LDA are too short
- weak bonds are noticeably too short, for example the Ni-C bond in the Ni carbonyl Ni(CO)4, the bond between two magnesium atoms (which are closed shell systems), and the length of hydrogen bonds such as that in the water dimer HOH-OH2;
- the binding energies calculated with the LDA are typically too large, sometimes by as much as 50% in strongly bound systems and even more in weakly bound materials.
Gradient-corrected density functionals as suggested by Perdew [19], Becke [20], Perdew & Wang [21] and Perdew, Burke & Ernzerhof [22] offer a remedy. The basic idea in these schemes is the inclusion of terms in the exchange-correlation expressions that depend on the gradient of the electron density and not only on its value at each point in space. Therefore, these corrections are also sometimes referred to as “non-local” potentials. The table below gives the form suggested by Becke175 for the exchange part and Perdew for the correlation.
While dissociation energies calculated with these corrections rival in accuracy the best post-Hartree-Fock quantum chemistry methods, gradient corrected density functional calculations are computationally much less demanding and more general. Gradient corrected density functionals have been studied extensively for molecular systems, for example by Andzelm & Wimmer [23] The results are very encouraging and this approach could turn out to be of great value in providing quantitative thermochemical data.
The one-particle eigenvalues obtained from gradient-corrected exchange-correlation potentials are not significantly different from the LDA eigenvalues. Therefore, these potentials do not (and are not intended to) remove the discrepancy between calculated and measured energy band gaps.
Gradient-correction to the total energy for exchange by Becke and correlation by Perdew
Energies are given in Hartree atomic units; the units for the electron and spin densities are number of electrons / (Bohr radius)3. The constant b in Becke’s formula is a parameter fitted to the exchange energy of inert gases. The explicit form of the functions f and g in Perdew’s expression for the correlation energy is given in the original paper.
3.1.4.1. Relativistic Effects
Electrons near an atomic nucleus achieve such high kinetic energies that relativistic effects become noticeable even for light atoms at the beginning of the periodic table. For elements with an atomic number above about 54 (Xe) these relativistic effects become quite important and should be included in electronic structure calculations. The relativistic mass enhancements of electrons near the nuclei lead to a contraction of the electronic charge distribution around the nuclei compared with a non-relativistic treatment. For atoms with about Z>54 non-relativistic calculations therefore can overestimate bond lengths by 0.1 \({\mathring{\mathrm{A}}}\) and more. Furthermore, relativistic effects change the relative energy of s, p, d, and f-states, which can have a significant impact on bonding mechanisms and energies.
Relativistic effects lead to a spin-orbit splitting which is, for example, about 0.3 eV for the splitting between the \(4f_{5/2}\) and \(4f_{7/2}\) in Ce. For core states, the spin orbit splitting can be very large. For example, the \(2p_{1/2}\) and \(2p_{3/2}\) core states in W are split by 1351 eV. Important effects on surfaces such as the Kerr rotation in magneto-optical devices or the x-ray dichroism involve spin-orbit splitting. Thus, a relativistic electronic structure theory is necessary. This is accomplished by solving the Dirac equations, as discussed, for example, in the textbooks by Bjorken & Drell [24] and Messiah. Within a spherically symmetric potential, the Dirac equations, like the non-relativistic Schrödinger equation, can be decomposed into a radial and angular part. For illustration, we show the radial equations
F and G are called the large and small component of the radial wave function. The quantum numbers n and l are equivalent to the non-relativistic case; j labels spin-orbit-split states and is used as subscript to label states such as \(2p_{\tfrac{1}{2}}\), \(2p_{\tfrac{3}{2}}\) and the \(4f_{\tfrac{5}{2}}\). The quantum number \(\kappa\) is a convenient quantity used within relativistic computer programs. The radial part of the charge density is constructed from the large and small components by
The correct treatment of exchange and correlation in a fully relativistic theory is a difficult problem and has not yet been completely resolved. However, reasonable approximations are available.
Koelling & Harmon [25] proposed a semi-relativistic (or scalar-relativistic) treatment. This approach involves an averaging over the spin-orbit splitting, but retains the kinematic effects. This restores most of the simplicity of a non-relativistic method, but still gives an excellent representation of the core electron distribution and the appropriate (spin-orbit averaged) energies of the valence electrons.
3.1.5. Atomic partial charges and Bader Charge Analysis
The concept of atomic charges, partial charges, angular momentum and crystal field split charge projections, and charge transfer is heavily used for describing and understanding chemical bonding and reactions, not only for molecular but also for surface and bulk systems. In order to address these needs, different approaches are available from MedeA-Vasp, which are briefly outlined here.
The definition of atomic charges is to some extent ambiguous, mainly due to the spatial continuity of the electron density distribution, the quantum-mechanical uncertainty of the electron position, and the difficulty to delimit between one atom and the neighboring one. Therefore, a quantitative analysis is usually only possible with respect to a suitable reference system, such as bulk vs. surface, or bound vs. isolated atom etc.
In particular the method used to partition space into parts attributed to each atom and an interstitial part strongly affects the quantitative results of the charge analysis. Most straightforwardly, space is partitioned into spheres of different radii around each atom position and the space in between. The Vasp code considers such a partitioning, and applies two different algorithms to compute atomic charges and charge contributions from different angular momentum channels (s, p, d, f partial charges) inside the spheres:
i) Projections P of the wave functions \(f_{nk}\) onto a set of spherical harmonics \(Y_{lm}\) centered at each atomic site N (which represents a natural basis set for atomic-like wave functions)
and the corresponding augmentation part. For this projection scheme the atomic sphere radii can be freely chosen, and as a default MedeA applies covalent radii for this purpose, which could be modified by setting the RWIGS parameter in the Add to Input Tab of the MedeA VASP GUI.
ii) Alternatively, a fast projection scheme onto the PAW spheres is implemented, for which the radii cannot be freely chosen but are fixed to the PAW sphere radii for each atom.
Further details on applying these projection schemes are provided in the section on the DOS/Optic/Tensors Tab of the MedeA VASP GUI and in the context sensitive Help panel.
The above projection schemes are based on somewhat arbitrary sphere radii, which strongly affect the charge data. Attempting to overcome this limitation, a number of schemes for charge analysis have been suggested, some are based on a population analysis of the wave functions applicable when basis functions centered at the atoms are used (Mulliken charges, Coulson charges), and some are based on a partitioning of the electron density distribution and are independent of the type of basis functions, therefore (Bader charges, Hirshfeld charges, Voronoi deformation density).
The MedeA VASP GUI offers in an automated fashion access to a Bader decomposition of space into areas attributed to each atom [26]. The definition where to delimit an atom is purely based on the total charge density as computed by Vasp. The division between atoms are made up by so-called zero flux surfaces S, i.e. surfaces where the electron density r satisfies the zero-flux boundary condition
for each point \(r_{s}\) of the surface, where \(n(r_{s})\) is the unit vector normal to the surface at \(r_{s}\). At each point of the dividing surfaces the gradient of the electron density has no component normal to the surface, i.e. the charge density exhibits a minimum perpendicular to the surface. Such surfaces of minimal charge density are intuitive and natural areas to separate atoms from each other. The regions bounded by these dividing surfaces are called Bader regions, which in most cases contain a nucleus, but it is also possible that no nucleus is found in a Bader region. Algorithmically, the Bader regions are identified by evaluating paths of steepest ascent confined to each grid point used for representing the charge density. Those grid points that have paths ending up at the same terminal maximum charge belong to the same Bader region. The total charge within a Bader region is evaluated by the sum over all these grid points, and provides a very good measure of the total electronic charge of an atom, the Bader charge.
The spatial extension of atoms - as arbitrarily defined by custom spheres or PAW spheres, or as identified as Bader regions from an analysis of the total charge density - is not only used to attribute total charges, s, p, d, and f charge characters and magnetic moments, but also for calculation and display of atom and angular momentum projected densities of states.
3.1.6. Implicit Solvation Model
As of version 2.19, MedeA VASP enables to simulate the effects of solvation in calculations for molecules, surfaces, adsorption processes and surface reactions. Solvation effects can be included for single point total energies, geometry optimizations and molecular dynamics, but making use of MedeA Phonon also vibrational properties of surface processes under solvation, and applying MedeA Transition State Search the energy profiles and reaction barriers in the presence of a solvent can be analyzed. The method called VASPsol was implemented in the group of Richard Hennig at University of Florida [27].
There are two main approaches for studying solvation effects by means of quantum theory. One approach treats the entire solute/solvent system in an atomistic and quantum mechanical manner, which extends the ab-initio approach explicitly to all solvent molecules. Due to the large number of solvent molecules required and the huge number of configurations needed for converged averaged equilibrium properties, such an explicit approach is computationally very expensive. The alternative approach, as pursued by VASPsol, treats only the solute system by the ab initio approach, which is in contact with a solvent whose properties are simulated by means of a continuum approach. The average over the solvent degrees of freedom are implicitly treated by the properties of the solvent bath. Such an implicit solvation model is computationally much more feasible and can be quite accurate, provided all interactions between solute and solvent are considered with appropriate detail.
There are three contributions taken into account by the VASPsol approach: The most important contribution, in particular for polar or ionic solutes or surfaces and polar solvents, is the electrostatic interaction, which is triggered by the dielectric constant as an input parameter. If both the solute/surface and the solvent is non-polar, the dispersive (Van der Waals) interactions might become more important than electrostatics. Finally, if the solvent molecules are quite large, the energy for creating a cavity within the solvent may become the dominant term, which is triggered by a surface tension parameter.
For the details of the derivation of the solvation model we refer to the publication of the VASPsol implementation, here just the basic assumptions and approximations are outlined. As a starting point, the free energy A of the combined solute/solvent system is written as the sum of a functional F of the total electron density and the thermodynamically averaged atomic densities of the solvent molecules and a term describing the electrostatic energy
with the total electron density being the sum of the electron density of the solute and the solvent \(n_{tot}(r) = n_{solute}(r) + n_{solvent}(r)\), \(N_{i}(r)\) denoting the thermodynamically averaged atomic densities of the solvent species i, and \(V_{ext}(r)\) being the external potential of the solute nuclei. The ground state free energy \(A_{0}\) is determined by a stepwise minimization of this functional, first over the solvent electron density and then over the solute electron density. After a separation into the usual Kohn-Sham density functional \(A_{KS}\) for the solute and the remaining term \(A_{diel}\) capturing all the interactions of the solute with the solvent as well as the internal energy of the solvent, minimization of \(A_{diel}\) with respect to the average atomic densities of the solvent \(N_{i}(r)\) finally yields the ground state free energy as
which is a functional of the electron density and the external potential of the solute only. All solvent effects are cast into the functional \(\tilde{A}_{diel}\) obtained from minimization over the solvent charge density and the average atomic densities of the solvent, thus representing a continuum model for the solvent, with its ground state being determined by the solute electronic structure. The expression (1.37) is exact, however, approximations for \(\tilde{A}_{diel}\) are needed to enable practical calculations.
One approximation refers to electrostatic solute-solvent interactions, which is introduced by a term included into the functional \(\tilde{A}_{diel}\) to account for the electrostatic interaction between the solute electronic structure and the charge distribution induced in the solvent. Given a linear dependence of the solvent polarization on the electric field close to the solute, the solvent polarization can be described by the local relative permittivity, the local dielectric function, \(e(r)\), of the solvent. On the other hand, to account for cavitation and dispersion, which dominantly appears in the first solvation shell, an interface term proportional to the solvent accessible area is introduced into the functional \(\tilde{A}_{diel}\), i.e. the free energy contribution
where t is an effective surface tension parameter describing cavitation, dispersion and repulsion between solvent and solute not covered by electrostatics, and S(r) is a cavity shape function. Inside the dielectric cavity the relative permittivity is assumed to be the vacuum value \(\epsilon(r) = 1\), and outside the bulk value of the solvent is reached with the induced charges being placed at the surface. Furthermore, a diffuse cavity is assumed, i.e. the relative permittivity is smoothly varying as a functional of the electronic charge density of the solute, which ensures continuous derivatives of the energy functional. The following functional dependence of the relative permittivity of the solvent on the solute electronic charge density is assumed
where \(e_{b}\) is the relative permittivity (dielectric constant) of the bulk solvent and
is the cavity shape function. The parameter \(n_{c}\) defines the value of the electron density at which the cavity forms and the parameter \(\sigma\) determines the width of the diffuse cavity. Within the range given by \(\sigma\) the relative permittivity is ramped up smoothly from the vacuum value of 1 inside the solute to \(\varepsilon_{b}\) inside the bulk solvent, and for \(\varepsilon_{b} \rightarrow 1\) the free energy becomes the vacuum value. The smooth variation mimics the first solvation shell, where the relative permittivity is known to be smaller than in the bulk due to the higher electric field near the solute (dielectric saturation).
Based on these approximations to electrostatic, cavitational and dispersive interactions the self-consistent solution of the Kohn-Sham equations for the solute/solvent system has become computationally practicable. From the solute charge density the combined electrostatic potential due to the electronic and nuclear charges of the solute in a polarizable medium is obtained by iteratively solving a generalized Poisson equation
where \(N_{solute}(r)\) are the effective core charges approximated by Gaussians and \(n_{solute}(r)\) is the valence electronic charge density of the solute. The Kohn-Sham Hamiltonian exhibits two additional terms resulting from solvation, an electrostatic and a cavitation term
The resulting energy expression has two additional related contributions covering electrostatic interactions and cavitation
In addition, the Hellman-Feynman force expression needs two correction terms due to solvation.
The parameters of this implicit solvation model can be set as input parameters of the VASP code in the INCAR file. The most prominent parameter, which is usually known from experimental data, is the bulk relative permittivity (or dielectric constant) of the solvent, \(\varepsilon_{b}\), which correspond to the EB_K keyword of VASP and can be set directly in the MedeA VASP GUI. The further parameters, i.e. \(n_{c}\) corresponding to NC_K, S corresponding to SIGMA_K, and \(\tau\) corresponding to the TAU keyword, can be set from the Add to Input tab, but are not accessible from experimental data, unfortunately. The default parameters are suitable for water: the dielectric constant \(\varepsilon = 78.4\) is set from experiment, whereas the shape function parameters \(n_{c}\) and \(\sigma\) and the effective surface tension \(t\) are obtained from a fit to experimental data on solvation energies of molecules in water: \(n_{c} = 0.0025\) \({\mathring{\mathrm{A}}}\)-3, \(\sigma = 0.6\), and \(t = 0.525\) meV \({\mathring{\mathrm{A}}}\)-2.
| [1] | Pierre Hohenberg and Walter Kohn, “Inhomogeneous Electron Gas,” Physical Review B 136, no. 3 (1964): 864-871. |
| [2] | Walter Kohn and L J Sham, “Self Consistent Equations Including Exchange and Correlation Effects,” Physical Review A 140, no. 4 (1965): 1133-1138. |
| [3] | L H Thomas, “The Calculation of Atomic Fields,” Proc. Cambridge Philos. Soc. 23 (1927): 542-548. |
| [4] | E Fermi, “Eine Statistische Methode Zur Bestimmung Einiger Eigenschaften Des Atoms Und Ihre Anwendung Auf Die Theorie Des Periodischen Systems,” Zeitschrift F r Physik 48 (1928): 73-79. |
| [5] | D R Hartree, “The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part I. Theory and Methods,” Proc. Cambridge Philos. Soc. 24 (1928): 89-110; D R Hartree, “The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part II. Some Results and Discussion,” Proc. Cambridge Philos. Soc. 24 (1928): 111-132; Vladimir Fock, “Näherungsmethode Zur Lösung Des Quantenmechanischen Mehrkörperproblems,” Zeitschrift F r Physik 61, no. 1 (January 1930): 126-148; Vladimir Fock, “‘Selfconsistent Field’ Mit Austausch F r Natrium,” Zeitschrift F r Physik 62, no. 11 (November 1930): 795-805. |
| [6] | J C Slater, “A Simplification of the Hartree-Fock Method,” Physical Review 81, no. 3 (February 1951): 385-390. |
| [7] | J C Slater, Quantum Theory of Molecules and Solids, McGraw-Hill, 1963. |
| [8] | L Hedin and B I Lundqvist, “Explicit Local Exchange-Correlation Potentials,” Journal of Physics C: Solid State Physics 4, no. 14 (March 14, 2001): 2064-2083. U von Barth and L Hedin, “A Local Exchange-Correlation Potential for the Spin Polarized Case. I,” Journal of Physics C: Solid State Physics, 1972. |
| [9] | D M Ceperley, “Ground State of the Electron Gas by a Stochastic Method,” Physical Review Letters 45, no. 7 (August 1980): 566-569. |
| [10] | ReszöGáspár, “Über Eine Approximation Des Hartree-Fockschen Potentials Durch Eine Universelle Potentialfunktion,” Acta Physica Academiae Scientiarum Hungaricae 3, no. 3 (April 1954): 263-286. |
| [11] | Walter Kohn and L J Sham, “Self Consistent Equations Including Exchange and Correlation Effects,” Physical Review A 140, no. 4 (1965): 1133-1138. |
| [12] | L Hedin and B I Lundqvist, “Explicit Local Exchange-Correlation Potentials,” Journal of Physics C: Solid State Physics 4, no. 14 (March 14, 2001): 2064-2083. |
| [13] | J Janak, “Proof That \({\delta}\) E/\({\delta}\) Ni=E in Density-Functional Theory,” Physical Review B 18, no. 12 (December 15, 1978): 7165. |
| [14] | Mark Hybertsen and Steven Louie, “First-Principles Theory of Quasiparticles: Calculation of Band Gaps in Semiconductors and Insulators,” Physical Review Letters 55, no. 13 (September 23, 1985): 1418. |
| [15] | F Schulte, “On the Theory of the Work Function,” Zeitschrift F r Physik B Condensed Matter 27, no. 4 (1977): 303-307. |
| [16] | U von Barth and L Hedin, “A Local Exchange-Correlation Potential for the Spin Polarized Case. I,” Journal of Physics C: Solid State Physics, 1972. |
| [17] | O Gunnarson, BI Lundqvist, and S Lundqvist, “Screening in a Spin-Polarized Electron Liquid,” Solid State Communications 11, no. 1 (1972): 149-153. |
| [18] | U von Barth and L Hedin, “A Local Exchange-Correlation Potential for the Spin Polarized Case. I,” Journal of Physics C: Solid State Physics, 1972. |
| [19] | John P Perdew, “Density Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas,” Physical Review B 33, no. 12 (1986): 8822-8824. |
| [20] | A D Becke, “Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior,” Physical Review A 38, no. 6 (1988): 3098. |
| [21] | John P Perdew and Yue Wang, “Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy,” Physical Review B 45, no. 23 (June 1992): 13244-13249. |
| [22] | John P Perdew, Kieron Burke, and Matthias Ernzerhof, “Generalized Gradient Approximation Made Simple,” Physical Review Letters 77, no. 18 (October 1996): 3865-3868. |
| [23] | J Andzelm and Erich Wimmer, “Density Functional Gaussian-Type-Orbital Approach to Molecular Geometries, Vibrations, and Reaction Energies,” Journal of Physical Chemistry 96, no. 2 (1992): 1280. |
| [24] | JD Bjorken and SD Drell, Relativistic Quantum Mechanics, McGraw-Hill, 1964. JD Bjorken and SD Drell, Relativistic Quantum Fields, McGraw-Hill, 1965. |
| [25] | D D Koelling and B N Harmon, “A Technique for Relativistic Spin-Polarised Calculations,” Journal of Physics C: Solid State Physics, 1977. |
| [26] | W. Tang, E. Sanville, and G. Henkelman, “A grid-based Bader analysis algorithm without lattice bias”, Journal of Physics: Condensed Matter 21, (2009): 084204. E. Sanville, S. D. Kenny, R. Smith, and G. Henkelman, “An improved grid-based algorithm for Bader charge allocation”, Journal of Computational Chemistry 28, (2007): 899-908. G. Henkelman, A. Arnaldsson, and H. Jónsson, “A fast and robust algorithm for Bader decomposition of charge density”, Computational Materials Science 36, (2006): 254-360 |
| [27] | K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T.A. Arias, R.G. Hennig, “Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways”, Journal of Chemical Physics 140, (2014): 084106 |
| download: | pdf |
|---|