2.19. Forcefields
Contents
| download: | pdf |
|---|
2.19.1. Selecting a Forcefield
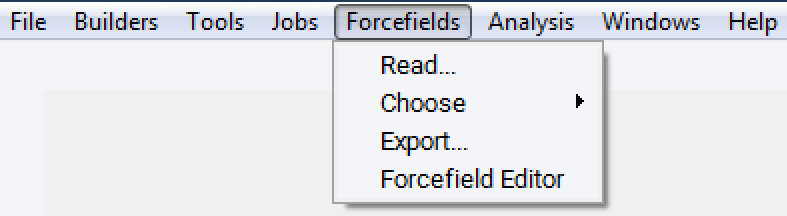
To select a specific forcefield, read in a forcefield file from Forcefields >> Read… .
One forcefield file can contain different versions, and most likely you want to use the default version.
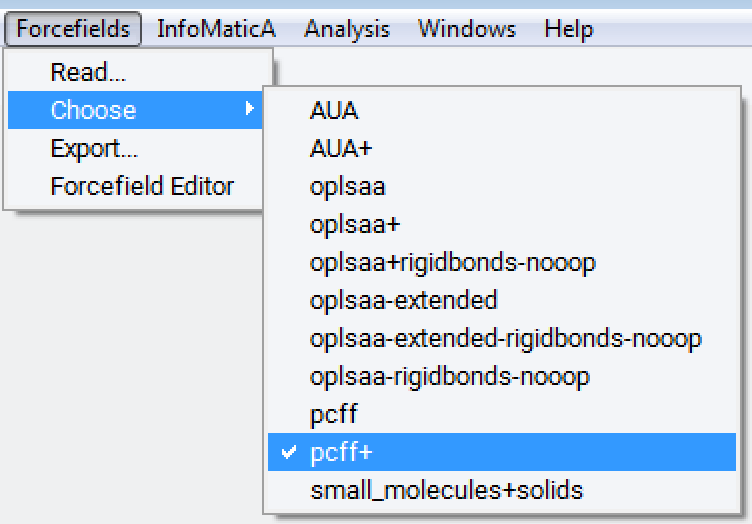
You can select or verify using a specific variant via Forcefield >> Choose. In this case you want to make sure that you use the additions from MaterialsDesign to pcff:
The initial selection comprises forcefields for:
Organic molecules and polymers
|
|
Inorganic compounds
|
|
Semiconductors
|
|
Nist Interatomic Potentials Repository
|
|
Noble gases
argon_rahman.frc
ReaxFF forcefields
|
|
2.19.2. Assigning Forcefield Parameters and Charges
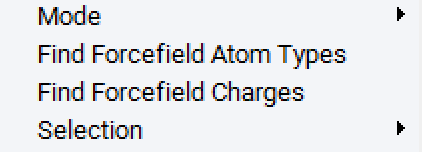
Right-click in your model window and use Find Forcefield Atom types and Find Forcefield Charges to perform the automatic atom type and partial charge assignment.
Note that when using covalent forcefields, it is important to ensure that appropriate bonds and bond orders - single, double, partial-double, triple etc. - have been set in order for correct atom types to be assigned using the forcefield’s atom template definitions. Failure to use correct bond orders generally results in atoms with incorrect chemical valence, which can give the misleading impression that a forcefield cannot be used for a given molecule. Therefore, if attempts at atom type assignment result in a warning message indicating that the atom type assignment resulted in unknown atom types, you should first ensure that the chemical structures of the molecules in the model are correct.
You may inspect the assignment by clicking on the Spreadsheet Icon  , where
atom types are listed in the FF Atom Type column (with ‘?’ used to denote any unassigned
types). If necessary, the spreadsheet also allows you to change charges and assign any
atom type for a selected atom or group of atoms.
, where
atom types are listed in the FF Atom Type column (with ‘?’ used to denote any unassigned
types). If necessary, the spreadsheet also allows you to change charges and assign any
atom type for a selected atom or group of atoms.
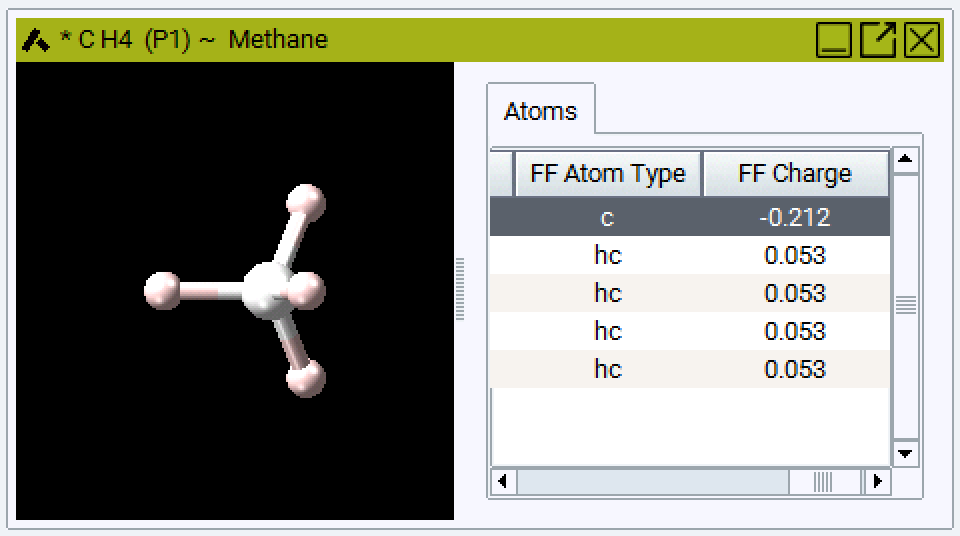
You can skip the automatic assignment and set all forcefield related values by hand (e.g. to match a publication). In this case, open the spreadsheet and you won’t see the columns FF Atom Type and FF Charge. Insert them by right-clicking in the heading of the spreadsheet and select new columns FF Atom Type and FF Charge.
You can arrange and sort the atoms in the model and assign atom types to groups by selecting more than one field: The selected fields are highlighted in blue, the active field is white. In the example below you can choose one atom type for all four H atoms in Methane.
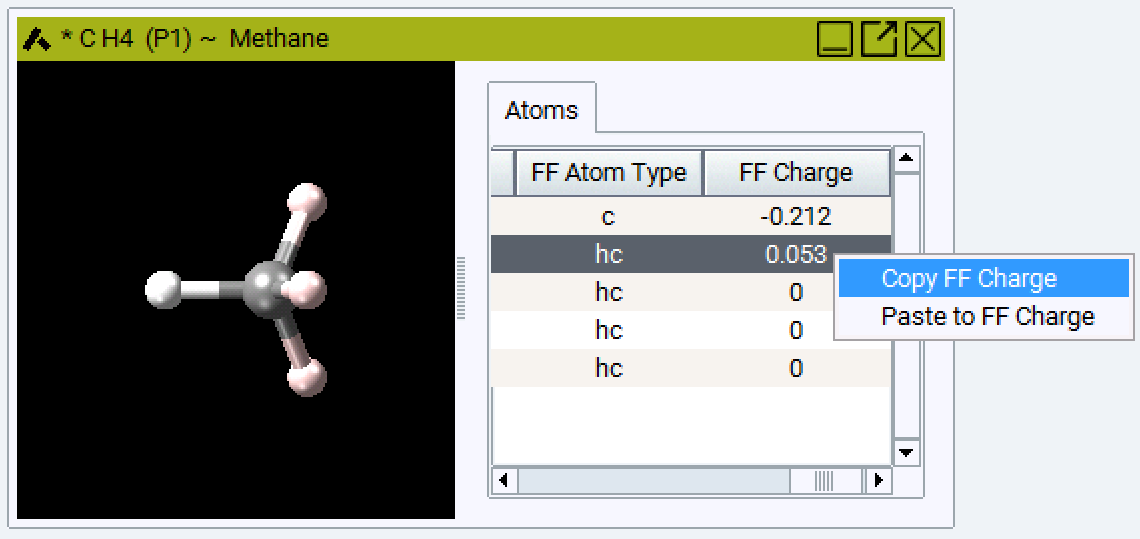
Setting charges is similar:
Enter charge for the first atom. Click into field and copy with right-click >> Copy FF charge
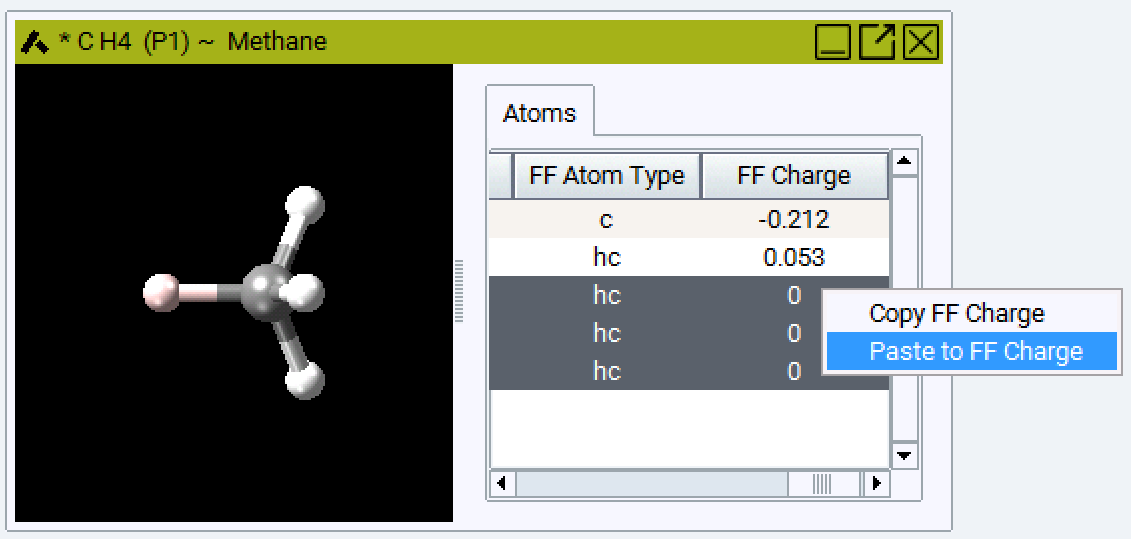
Select remaining atoms with mouse and paste with right-click >> Paste to FF charge
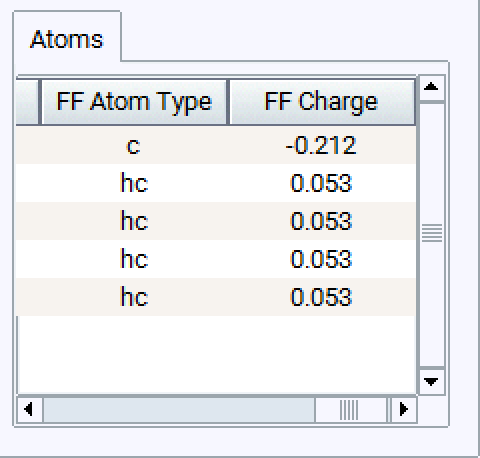
Charge values are copied into the selection
2.19.3. Forcefield Overview
2.19.3.1. Organic Molecules and Polymers
We recommend and support the use of pcff+ for all atom molecular dynamics, energy minimization, and related simulations. OPLS-AA, AUA and TraPPE are supplied to use with MedeA GIBBS for computationally efficient configurational space sampling and the use of extended atoms.
All the forcefields for organic systems require topological information (such as bonds and bond orders) to determine the atom type and charge for each atom. These forcefields cannot describe the creation or the breaking of bonds.
2.19.3.1.1. pcff+.frc:
A significant extension to the pcff.frc included with the LAMMPS distribution (see, for example, Sun, Mumby, Maple & Hagler [1]). pcff+.frc preserves the ‘cff-series’ ab-initio based parameters for valence interactions (as used in cff91.frc, cff93.frc and pcff.frc). This is supplemented by a substantial refinement of nonbonded parameters based on high quality experimental data for small molecule liquids and gases, together with new parameterizations for selected compounds such as thiophenes. Details are given in the References section at the end of the file.
| Ag | Ag | Silver metal |
| Al | Al | Aluminium metal |
| Au | Au | Gold metal |
| Br | Br | bromine ion |
| Cl | Cl | chlorine ion |
| Cr | Cr | Chromium metal |
| Cu | Cu | Copper metal |
| Fe | Fe | Iron metal |
| K | K | Potassium metal |
| Li | Li | Lithium metal |
| Mo | Mo | Molybdenum metal |
| Na | Na | Sodium metal |
| Ni | Ni | Nickel metal |
| Pb | Pb | Lead metal |
| Pd | Pd | Palladium metal |
| Pt | Pt | Platinum metal |
| Sn | Sn | Tin metal |
| W | W | Tungsten metal |
| Ar | ar | Argon |
| Al | az | aluminium atom in zeolites |
| Br | br | bromine atom |
| C | c | generic SP3 carbon |
| C | c+ | C in guanidinium group |
| C | c- | C in charged carboxylate |
| C | c1 | sp3 carbon with 1 H 3 heavies |
| C | c2 | sp3 carbon with 2 H’s, 2 Heavy’s |
| C | c3 | sp3 carbon with 3 hHs 1 heavy |
| C | c3h | sp3 carbon in 3-membered ring with hydrogens |
| C | c3m | sp3 carbon in 3-membered ring |
| C | c4h | sp3 carbon in 4-membered ring with hydrogens |
| C | c4m | sp3 carbon in 4-membered ring |
| C | c5 | sp2 aromatic carbon in 5-membered ring |
| C | c= | non aromatic end doubly bonded carbon |
| C | c=1 | non aromatic, next to end doubly bonded carbon |
| C | c=2 | non aromatic doubly bonded carbon |
| C | c_0 | carbonyl carbon of aldehydes, ketones |
| C | c_1 | carbonyl carbon of acid, ester, amide |
| C | c_2 | carbonyl carbon of carbamate, urea |
| C | c_a | general amino acid alpha carbon (sp3) |
| Ca | ca+ | calcium ion |
| C | cg | sp3 alpha carbon in glycine |
| C | ci | sp2 aromatic carbon in charged imidazole ring (His+) |
| Cl | cl | chlorine atom |
| C | co | sp3 carbon in acetals |
| C | coh | sp3 carbon in acetals with hydrogen |
| C | cp | sp2 aromatic carbon |
| C | cr | C in neutral arginine |
| C | cs | sp2 aromatic carbon in 5 membered ring next to S |
| C | ct | sp carbon involved in a triple bond |
| C | cz | carbonyl carbon of carbonate |
| D | dw | deuterium in heivy water |
| F | f | fluorine atom |
| H | h | generic hydrogen bound to C, Si,or H |
| H | h* | hydrogen bonded to nitrogen, Oxygen |
| H | h+ | charged hydrogen in cations |
| H | hb | hydrogen atom in bridging hydroxyl group |
| H | hc | hydrogen bonded to carbon |
| He | he | Helium |
| H | hi | Hydrogen in charged imidazole ring |
| H | hn | hydrogen bonded to nitrogen |
| H | hn2 | amino hydrogen |
| H | ho | hydrogen bonded to oxygen |
| H | ho2 | hydroxyl hydrogen |
| H | hoa | hydrogen atom in terminal hydroxyl group on aluminium |
| H | hos | hydrogen atom in terminal hydroxyl group on silicon |
| H | hp | hydrogen bonded to phosphorus |
| H | hs | hydrogen bonded to sulfur |
| H | hsi | silane hydrogen |
| H | hw | hydrogen in water |
| I | i | iodine atom |
| Kr | kr | Krypton |
| N | n | generic sp2 nitrogen (in amids)) |
| N | n+ | sp3 nitrogen in protonated amines |
| N | n1 | sp2 nitrogen in charged arginine |
| N | n2 | sp2 nitrogen (NH2) in guanidinium group (HN=C(NH2)2) |
| N | n3m | sp3 nitrogen in 3- membered ring |
| N | n3n | sp2 nitrogen in 3- membered ring |
| N | n4 | sp3 nitrogen in protonated amines |
| N | n4m | sp3 nitrogen in 4- membered ring |
| N | n4n | sp2 nitrogen in 4- membered ring |
| N | n= | non aromatic end doubly bonded nitrogen |
| N | n=1 | non aromatic, next to end doubly bonded carbon |
| N | n=2 | non aromatic doubly bonded nitrogen |
| N | n_2 | nitrogen of urethane |
| N | na | sp3 nitrogen in amines |
| N | nb | sp2 nitrogen in aromatic amines |
| Ne | ne | Neon |
| N | nh | sp2 nitrogen in 5 or 6 membered ring |
| N | nh+ | protonated nitrogen in 6 membered ring |
| N | nho | sp2 nitrogen in 6 membered ring next to a carbonyl |
| N | ni | nitrogen in charged imidazole ring |
| N | nn | sp2 nitrogen in aromatic amines |
| N | np | sp2 nitrogen in 5- or 6- membered ring |
| N | npc | sp2 nitrogen in 5- or 6- membered ring and with a heavy atom |
| N | nr | sp2 nitrogen (NH2) in guanidinium group (HN=C(NH2)2) |
| N | nt | sp nitrogen involved in a triple bond |
| N | nz | sp3 nitrogen bonded to two atoms |
| O | o | generic SP3 oxygen |
| O | o* | oxygen in water |
| O | o- | partial double oxygen |
| O | o3e | sp3 oxygen in three membered ring |
| O | o4e | sp3 oxygen in four membered ring |
| O | o= | oxygen double bonded to O, C, S, N, P |
| O | o_1 | oxygen in carbonyl group |
| O | o_2 | ester oxygen |
| O | oah | oxygen atom in terminal hydroxyl group on aluminium |
| O | oas | oxygen atom between aluminium and silicon |
| O | ob | oxygen atom in bridging hydroxyl group |
| O | oc | sp3 oxygen in ether or acetals |
| O | oe | sp3 oxygen in ester |
| O | oh | oxygen bonded to hydrogen |
| O | oo | oxygen in carbonyl group, carbonate only |
| O | op | sp2 aromatic in 5 membered ring |
| O | osh | oxygen atom in terminal hydroxyl group on silicon |
| O | osi | siloxane oxygen |
| O | oss | oxygen atom betweem two silicons |
| O | oz | ester oxygen in carbonate |
| P | p | general phosphorous atom |
| P | p= | phosphazene phosphorous atom |
| S | s | sp3 sulfur |
| S | s’ | S in thioketone group |
| S | s- | partial double sulfur |
| S | s1 | sp3 sulfur involved in (S-S) group of disulfides |
| S | s3e | sulfur in three membered ring |
| S | s4e | sulfur in four membered ring |
| S | sc | sp3 sulfur in methionines (C-S-C) group |
| S | sf | S in sulfonate group |
| S | sh | sp3 sulfur in sulfhydryl (-SH) group (e.g. cysteine) |
| Si | si | silicon atom |
| Si | sio | siloxane silicon |
| S | sp | sulfur in an aromatic ring (e.g. thiophene) |
| Si | sz | silicon atom in zeolites |
| Xe | xe | Xenon |
| As | as | Arsenic in AsR3 |
| B | b3n | sp2 boron in hexagonal boron nitride |
| Br | brh | bromine in HBr molecule |
| C | c0 | sp3 carbon with 0 H, 4 heavies |
| C | c0x | sp3 carbon with 0 H, 4 fluorines |
| C | c1o | carbon in CO |
| C | c2= | carbon in CO2 and CS2 |
| C | c3as | sp3 carbon in methyl arsines |
| C | c3h1 | sp3 carbon in 3-membered ring with one hydrogen |
| C | c3si | sp3 carbon with 3 hydrogens and Si |
| C | c3o- | carbon in carbonate anion |
| C | c41o | carbon, sp3, in methanol |
| C | c43o | carbon, sp3 in secondary alcohols |
| C | c4h1 | sp3 carbon in 4-membered ring with one hydrogen |
| C | c4o | alpha carbon |
| C | c0oe | alpha carbon in ether containing tertiary alkyl group, e.g. -C-O-C-R3 |
| C | c1oe | alpha carbon in ether containing secondary alkyl group, e.g. -C-O-CH-R2 |
| C | c2oe | alpha carbon in ether containing primary alkyl group, -C-O-CH2-R |
| C | c2oz | alpha carbon in carbonates -O(O)C-O-CH2-R |
| C | c3oe | alpha carbon in methyl containing ethers -C-O-CH3 |
| C | c3oz | alpha carbon in methyl-containing carbonates -O(O)C-O-CH3 |
| C | c4oe | alpha carbon in general ethers -C-O-C- (legacy) |
| C | c5h | sp3 carbon in 5-membered ring |
| C | c5h1 | sp3 carbon in 5-membered ring with one hydrogen |
| Cl | cl4 | chlorine in ClO4- anion |
| Cl | clh | chlorine in HCl molecule |
| C | cpc | alpha/ipso carbon in aromatic ethers -C-O-C- |
| Cs | Cs+ | cesium ion |
| F | ff | fluorine atom in perfluorinated aliphatics |
| F | ffp | fluorine atom in perfluorinated aromatics |
| F | F | fluorine ion |
| Ge | ge4 | generic germanium with four bonds attached |
| H | h1h | hydrogen in H2 |
| H | h_1p | hydrogen in NH4+ |
| H | hbr | hydrogen in HBr molecule |
| H | hcl | hydrogen in HCl molecule |
| H | hhi | hydrogen in HI molecule |
| H | ho- | hydrogen in hydroxide ion OH- |
| I | I | iodine ion |
| I | ih | iodine in HI molecule |
| K | K+ | potassium ion |
| Li | Li+ | lithium ion |
| N | n1o | nitrogen in NO |
| N | n2o | nitrogen in NO2 |
| N | n2- | nitrogen in amide/imide anion |
| N | n3b | sp2 nitrogen in hexagonal boron nitride |
| N | n4o | nitrogen in amine oxides |
| N | n_3 | nitrogen in primary or secondary amide |
| N | n_3- | nitrogen in NO3- nitrate ion |
| N | n_30 | nitrogen in tertiary amide |
| N | n_31 | nitrogen in secondary amide |
| N | n_32 | nitrogen in primary amide |
| N | n_4 | nitrogen in NH4+ |
| N | n_4c | nitrogen in NR4+ |
| N | na0 | sp3 nitrogen in tertiary aliphatic amines |
| N | na1 | sp3 nitrogen in secondary aliphatic amines |
| N | na2 | sp3 nitrogen in primary aliphatic amines (same as na) |
| N | nbo | sp2 nitrogen in aromatic nitro compounds |
| Na | Na+ | sodium ion |
| O | o=n | oxygen double bonded to N in aromatic nitro group |
| O | o1= | oxygen in NO2 and SO2 |
| O | o1=* | oxygen in CO2 |
| O | o1c | oxygen in CO |
| O | o1c- | oxygen in carbonate anion |
| O | o1n | oxygen in NO |
| O | o1n4 | oxygen in amine oxides |
| O | o1o | oxygen in O2 |
| O | o1s- | oxygen in sulfate or sulfonate anion |
| O | o1n- | oxygen in nitrate ion |
| O | o2s- | ether oxygen in sulfate anion |
| O | o_1h | oxygen in carbonyl group of aldehydes |
| O | o_1r | oxygen in ClO4- anion |
| O | o_2c | oxygen in carboxylic acids |
| O | oc | sp3 oxygen in ether or acetals |
| O | oh- | oxygen in hydroxide ion OH- |
| P | p6- | phosphorous in phosphate |
| P | ph3 | phosphorous in phosphine |
| Rb | Rb+ | rubidium ion |
| S | s1= | sulfur in CS2 |
| S | s2= | sulfur in SO2 |
| S | se- | sulfur in sulfate anion |
2.19.3.1.2. oplsaa+.frc
Based on Jorgensen, Maxwell & Tirado-Rives [2] (oplsaa), supplemented with inclusion of additional parameters derived by various groups (oplsaa_extended), and original work by Materials Design (oplsaa+).
| Ar | Ar | Argon atom |
| C | C | Carbonyl carbon in amides, esters |
| C | CA | Aromatic carbon |
| C | CAh1 | Aromatic carbon pyridine atom 2 |
| C | CAh2 | Aromatic carbon pyridine atom 3 |
| C | CAh3 | Aromatic carbon pyridine atom 4 |
| C | CAh4 | Aromatic carbon pyrimidine atom 3 |
| C | CAh5 | Aromatic carbon pyrimidine atom 4 |
| C | CAh6 | Aromatic carbon pyridazine atom 2 |
| C | CAh7 | Aromatic carbon pyridazine atom 3 |
| C | CAh8 | Aromatic carbon pyrazine |
| C | CAh9 | Aromatic carbon pyrazole |
| C | CAh0 | Aromatic carbon isoxazole |
| C | CAi1 | Aromatic carbon indole atom 4 |
| C | CAi2 | Aromatic carbon indole atom 5 |
| C | CAi3 | Aromatic carbon indole atom 6 |
| C | CAi4 | Aromatic carbon indole atom 7 |
| C | CB | Aromatic carbon indole atom 9 |
| C | CM | sp2 aliphatic carbon |
| C | CN | aromatic carbon indole atom 8 |
| C | CO | Acetal carbon ROCOR |
| C | CQ | pyrimidine N-C-N aromatic carbon |
| C | CR | Aromatic carbon imidazole |
| C | CRh1 | Aromatic carbon oxazole |
| C | CS | Generic 5-membered ring carbon |
| C | CSh1 | Aromatic carbon pyrrole |
| C | CSh2 | Aromatic carbon furan |
| C | CSh3 | Aromatic carbon indole atom 3 |
| C | CT | sp3 aliphatic carbon |
| C | CT1 | sp3 alpha carbon in nitriles |
| C | CTEX | Exocyclic sp3 aliphatic carbon in cyclic amine |
| C | CTfn | Perfluoroalkane carbon |
| C | CTf4 | Tetrafluoromethane carbon |
| C | CU | Aromatic carbon pyrazole |
| C | CUh1 | Aromatic carbon isoxazole |
| C | CV | Aromatic carbon imidazole |
| C | CVh1 | Aromatic carbon oxazole |
| C | CW | sp2 aliphatic carbon |
| C | CWh1 | Aromatic carbon pyrrole |
| C | CWh2 | Aromatic carbon furan |
| C | CWh3 | Aromatic carbon pyrazole |
| C | CWh4 | Aromatic carbon isoxazole |
| C | CWh5 | Aromatic carbon imidazole |
| C | CWh6 | Aromatic carbon oxazole |
| C | CWh7 | Aromatic carbon indole atom 2 |
| C | CZ | sp alkyl nitrile carbon |
| C | CZ1 | sp aryl nitrile carbon |
| F | F | Fluorine in perfluorinated hydrocarbons |
| H | H | Amide or amine H(N) hydrogen |
| H | HA | Aromatic hydrogen |
| H | HC | Hydrogen bonded to carbon |
| H | HC1 | Hydrogen bonded to carbon in methanol |
| H | HC2 | Hydrogen bonded to carbon in alkenes RH-C= and H2-C= |
| H | HC3 | Hydrogen bonded to carbon in ethers |
| H | HC4 | Hydrogen bonded to carbon next to NR2, NO2, or nitrile |
| H | HC5 | alpha alkoxy H in esters |
| H | HC6 | H on alpha carbon of aldehyde and ketone |
| He | He | Helium atom |
| H | HEX4 | Amine hydrogen in 4-membered cyclic amine (azetidine) |
| H | HEX5 | Amine hydrogen in 5-membered cyclic amine (pyrrolidine) |
| H | HEX6 | Amine hydrogen in 6-membered cyclic amine (piperidine) |
| H | HW | Hydrogen in TIP3P water |
| H | HO | Hydrogen bonded to O |
| H | HS | Hydrogen bonded to S in thiols |
| Kr | Kr | Krypton atom |
| N | N | Nitrogen in amides |
| N | N1 | Nitrogen in primary amides |
| N | N2 | Nitrogen in secondary amides |
| N | N3 | Nitrogen in tertiary amides |
| Ne | Ne | Neon atom |
| N | NA | Nitrogen in pyrrole |
| N | NAh2 | N-H Nitrogen in pyrazole |
| N | NAh3 | N-H Nitrogen in imidazole |
| N | NAh4 | N-H Nitrogen in indole (atom 1) |
| N | NB | Nitrogen in pyrazole |
| N | NBh1 | Nitrogen in isoxazole |
| N | NBh2 | Nitrogen in imidazole |
| N | NBh3 | Nitrogen in oxazole |
| N | NC | Nitrogen in pyridine and diazenes |
| N | NO | Nitrogen in nitroalkane |
| N | NT0 | Nitrogen in ammonia |
| N | NT | Nitrogen in primary amines |
| N | NT2 | Nitrogen in secondary amines |
| N | NT3 | Nitrogen in tertiary amines |
| N | NZ | Nitrogen in nitriles |
| O | O | Oxygen in amides |
| O | O1 | Oxygen in carboxylate esters |
| O | O2 | Oxygen in aldehydes |
| O | O3 | Oxygen in ketones |
| O | O4 | Oxygen in carboxylic acids RCOOH |
| O | OH | Oxygen in hydroxyl (OH) group |
| O | OH2 | Oxygen in hydroxyl (OH) group (diols) |
| O | OH3 | Oxygen in hydroxyl (OH) group (triols) |
| O | OH4 | Oxygen in hydroxyl (OH) group (RCOOH) |
| O | OH5 | Oxygen in hydroxyl (OH) group (phenol) |
| O | ON | Oxygen in nitro group |
| O | OS | Oxygen in ethers, including acetals |
| O | OS1 | Alkoxy oxygen in esters |
| O | OW | Oxygen in TIP3P water |
| S | S | Sulfur in sulfides and disulfides |
| S | SH | Sulfur in thiols |
| S | SH1 | Sulfur in H2S |
| Xe | Xe | Xenon atom |
| C | C1i | aliphatic carbon bonded to N in R4N+ |
| C | C2i | aliphatic carbon bonded to C1i in R4N+ |
| C | CTi | sp3 aliphatic carbon in ionic liquid |
| F | Fi | Fluorine in ionic liquid anion |
| H | H1 | Hydrogen bonded to C1 in R4N+ cation |
| N | N2i | Nitrogen bonded to S in triflimide anion |
| N | N4i | Nitrogen in R4N+ cation |
| O | OYi | Oxygen bonded to S in triflate |
| S | SY6i | Sulfur in bis triflimide |
2.19.3.1.3. Trappe+.frc
Martin [3] , Kamath [4] , Stubbs [5] , Wick [6] , Chen [7] , Wick [8] , Martin [9] , Lubna [10] , Maerzke [11]
| C | C | Aliphatic |
| C | CHx-aliphatic | Aliphatic |
| C | CH4-TraPPE-UA | Molecule CH4-TraPPE-UA |
| C | CH3-TraPPE-UA | Group CH3-TraPPE-UA- |
| C | CH2-TraPPE-UA | Group CH2-TraPPE-UA- |
| C | CH-TraPPE-UA | Group CH-TraPPE-UA- |
| C | C-TraPPE-UA | Group C-TraPPE-UA- |
| C | CH2-olef-TraPPE-UA | Group CH2-olef-TraPPE-UA= |
| C | CH-olef-TraPPE-UA | Group CH-olef-TraPPE-UA= |
| C | C-olef-TraPPE-UA | Group C-olef-TraPPE-UA= |
| C | CH-EA-TraPPE-UA | Group CH-EA-TraPPE-UA- for C bonded to O in Ethers and Alcohols |
| C | CH-(EA)-TraPPE-UA | Group CH-EA-TraPPE-UA- for C bonded to O in Ethers and Alcohols |
| C | C-EA-TraPPE-UA | Group C-EA-TraPPE-UA- for C bonded to O in Ethers and Alcohols |
| C | C-(EA)-TraPPE-UA | Group C-EA-TraPPE-UA- for C bonded to O in Ethers and Alcohols |
| C | C-arom-TraPPE-UA | Aromatic C-arom-TraPPE-UA carbon |
| C | C-l-arom-TraPPE-UA | Aromatic C-arom-TraPPE-UA carbon linking two rings in condensed units (naphthalene, indane, phenanthrene,.. ) |
| C | C-d-arom-TraPPE-UA | Aromatic C-arom-TraPPE-UA carbon linking two rings in diphenyl |
| C | CH-arom-TraPPE-UA | Aromatic C-arom-TraPPE-UA carbon with one hydrogen |
| C | CH-aldehyde-TraPPE-UA | C connected to O in aldehydes TraPPE-UA |
| C | CH-(aldehyde)-TraPPE-UA | C connected to O in aldehydes TraPPE-UA |
| C | C-ketone-TraPPE-UA | C connented to O in ketones TraPPE-UA |
| C | C-(ketone)-TraPPE-UA | C connented to O in ketones TraPPE-UA |
| C | CH2-cyc5-TraPPE-UA | Group CH2- in a 5-membered cyclic non-aromatic ring |
| C | CH2-cyc6-TraPPE-UA | Group CH2- in a 6-membered cyclic non-aromatic ring |
| C | CH-cyc-TraPPE-UA | Group CH- in a 5- or 6-membered cyclic non-aromatic ring |
| C | C-cyc-TraPPE-UA | Group C- in a 5- or 6-membered cyclic non-aromatic ring |
| H | H-OH-TraPPE-UA | Hydrogen bonded to O in OH groups |
| H | H(-OH)-TraPPE-UA | Hydrogen bonded to O in OH groups |
| H | HA | Aromatic hydrogen |
| H | UnitedH | ghost H (used also for aromatic in this version) |
| H | H-SH-TraPPE-UA | H bonded with S in thiols |
| H | H-pyrrole-TraPPE-UA | H bonded with N in pyrrole |
| N | N-pyridine-TraPPE-UA | Nitrogen in pyridine |
| N | N-pyrrole-TraPPE-UA | Nitrogen in pyrrole |
| N | N-arom-TraPPE-UA | Nitrogen in aromatic rings |
| O | O-OH-TraPPE-UA | Oxygen in hydroxyl (O-OH-TraPPE-UA) group |
| O | O(-OH)-TraPPE-UA | Oxygen in hydroxyl (O-OH-TraPPE-UA) group |
| O | O-ROR-TraPPE-UA | Oxygen in ethers |
| O | O(ROR)-TraPPE-UA | Oxygen in ethers |
| O | O-aldehydeketone-TraPPE-UA | Oxygen in aldehydes and ketones TraPPE-UA |
| O | O-(aldehydeketone)-TraPPE-UA | Oxygen in aldehydes and ketones TraPPE-UA |
| S | S | Sulfur |
| S | S-thiol-TraPPE-UA | Sulfur in thiols |
| S | S-sulfide-TraPPE-UA | Sulfur in sulfides |
| S | S-disulfide-TraPPE-UA | Sulfur in disulfides |
| S | S-thiophene-TraPPE-UA | Sulfur in thiophene |
2.19.3.1.4. compass+.frc - The Published Part of COMPASS
Supplied for consistency with the LAMMPS distribution. General use of this forcefield is deprecated, as the forcefield is not maintained. [12] Contains the collection of compass parameters in their original published form. compass+.frc includes subsequently published corrections.
2.19.3.1.5. Cvff.frc
Supplied for consistency with the LAMMPS distribution. General use of this forcefield is deprecated. [13]
2.19.3.1.6. Cff91.frc
Supplied for consistency with the LAMMPS distribution. General use of this forcefield is deprecated. [14]
2.19.3.1.7. Cff93.frc
Supplied for consistency with the LAMMPS distribution. General use of this forcefield is deprecated. [15]
2.19.3.2. Inorganic Compounds
We don’t make overall recommendations for inorganic forcefields, because the local coordination of inorganic systems varies widely, and the transferability of forcefield terms cannot be assumed from one compound to another. The scope and applicability of forcefields for inorganics are best discerned through reference to their original derivation. These forcefields don’t require bonds.
2.19.3.2.1. inorganic.frc
Compiled by Woodley, Battle, Gale & Catlow [16], Xia [17] for use in inorganic crystal structure prediction.
| Ag | Ag1+ | |
| Ag | Ag3+ | |
| Al | Al3+ | |
| Ba | Ba2+ | |
| Ca | Ca2+ | |
| Cd | Cd2+ | |
| Ce | Ce4+ | |
| Co | Co2+ | |
| Co | Co3+ | |
| Cr | Cr3+ | |
| Cu | Cu1+ | |
| Fe | Fe2+ | |
| Fe | Fe3+ | |
| Ge | Ge4+ | |
| K | K1+ | |
| La | La3+ | |
| Mg | Mg2+ | |
| Mn | Mn2+ | |
| Mn | Mn4+ | |
| Na | Na1+ | |
| Nb | Nb5+ | |
| Ni | Ni2+ | |
| O | O2- | |
| O | O12- | |
| O | O22- | |
| Pb | Pb1+ | |
| Po | Po4+ | |
| Pr | Pr3+ | |
| Rb | Rb1+ | |
| Si | Si4+ | |
| Sn | Sn4+ | |
| Sr | Sr2+ | |
| Ta | Ta2+ | |
| Tl | Tl3+ | |
| Ti | Ti3+ | |
| Ti | Ti4+ | |
| U | U2+ | |
| V | V2+ | |
| V | V3+ | |
| V | V4+ | |
| Y | Y3+ | |
| Zn | Zn2+ | |
| Zr | Zr2+ |
2.19.3.2.2. bks.frc
Derived by van Beest, Kramer & van Santen [18] to provide a description of structural and vibrational properties for framework structure materials based on two-body (i.e. without explicit angle terms).
| Al | Al | |
| O | O | |
| P | P | |
| Si | Si |
2.19.3.2.3. CVFF_aug.frc
This forcefield was developed by Behnam Vessal using a methodology similar to that employed by van Beest, to create a broad two-body (i.e. without explicit angle terms) description of framework structured materials able to support extra framework atoms. [19]
| H | h | Hydrogen bonded to C. Masses from CRC 1973/74 pages B-250. |
| H | d | General Deuterium |
| H | hn | Hydrogen bonded to N |
| H | ho | Hydrogen bonded to O |
| H | hp | Hydrogen bonded to P |
| H | hs | Hydrogen bonded to S |
| H | h* | Hydrogen in water molecule |
| H | h$ | Hydrogen atom for automatic parameter assignment |
| L | lp | Lone Pair |
| L | lp | Lone Pair |
| H | h+ | Charged hydrogen in cations |
| H | hc | Hydrogen bonded to carbon |
| H | hi | Hydrogen in charged imidazole ring |
| H | hw | Hydrogen in water |
| D | dw | Deuterium in heivy water |
| C | c | Sp3 aliphatic carbon |
| C | cg | Sp3 alpha carbon in glycine |
| C | c’ | Sp2 carbon in carbonyl (C=O) group |
| C | c* | Carbon in carbonyl group, non_amides |
| C | c” | Carbon in carbonyl group, non_amides |
| C | cp | Sp2 aromatic carbon (partial double bonds) |
| C | cr | Carbon in guanidinium group (HN=C(NH2)2) |
| C | c+ | C in guanidinium group |
| C | c- | Carbon in charged carboxylate (COO-) group |
| C | ca | General amino acid alpha carbon (sp3) |
| C | c3 | Sp3 carbon in methyl (CH3) group |
| C | cn | Sp3 Carbon bonded to N |
| C | c2 | Sp3 carbon bonded to 2 H’s, 2 heavy atoms |
| C | c1 | Sp3 carbon bonded to 1 H, 3 Heavy atoms |
| C | c5 | Sp2 aromatic carbon in five membered ring |
| C | cs | Sp2 carbon involved in thiophene |
| C | c= | Non aromatic end doubly bonded carbon |
| C | c=1 | Non aromatic, next to end doubly bonded carbon |
| C | c=2 | Non aromatic doubly bonded carbon |
| C | ct | Sp carbon involved in triple bond |
| C | ci | Sp2 aromatic carbon in charged imidazole ring (His+) |
| C | c$ | Carbon atom for automatic parameter assignment |
| C | co | Sp3 carbon in acetals |
| C | c3m | Sp3 carbon in 3-membered ring |
| C | c4m | Sp3 carbon in 4-membered ring |
| C | coh | Sp3 carbon in acetals with hydrogen |
| C | c3h | Sp3 carbon in 3-membered ring with hydrogens |
| C | c4h | Sp3 carbon in 4-membered ring with hydrogens |
| C | ci | Sp2 aromatic carbon in charged imidazole ring (His+) |
| N | n | Sp2 nitrogen with 1 H, 2 heavy atoms (amide group) |
| N | no | Sp2 nitrogen in nitro group |
| N | n2 | Sp2 nitrogen (NH2 in the guanidinium group (HN=C(NH2)2)) |
| N | np | Sp2 aromatic nitrogen (partial double bonds) |
| N | n3 | Sp3 nitrogen with three substituents |
| N | n4 | Sp3 nitrogen with four substituents |
| N | n= | Non aromatic end double bonded nitrogen |
| N | n=1 | Non aromatic, next to end doubly bonded carbon |
| N | n=2 | Non aromatic doubly bonded nitrogen |
| N | nt | Sp nitrogen involved in triple bond |
| N | nz | Sp nitrogen in N2 |
| N | n1 | Sp2 nitrogen in charged arginine |
| N | ni | Sp2 nitrogen in a charged imidazole ring (HIS+) |
| N | n$ | Nitrogen atom for automatic parameter assignment |
| N | na | Sp3 nitrogen in amines |
| N | n3m | Sp3 nitrogen in 3- membered ring |
| N | n4m | Sp3 nitrogen in 4- membered ring |
| N | n3n | Sp2 nitrogen in 3- membered ring |
| N | n4n | Sp2 nitrogen in 4- membered ring |
| N | nb | sp2 nitrogen in aromatic amines |
| N | nn | sp2 nitrogen in aromatic amines |
| N | npc | sp2 nitrogen in 5- or 6- membered ring bonded to a heavy atom |
| N | nh | sp2 nitrogen in 5-or 6- membered ring with hydrogen attached |
| N | nho | sp2 nitrogen in 6- membered ring next to a carbonyl group and with a hydrogen |
| N | nh+ | protonated nitrogen in 6- membered ring with hydrogen attached |
| N | n+ | sp3 nitrogen in protonated amines |
| N | nr | sp2 nitrogen (NH2) in guanidinium group (HN=C(NH2)2) |
| O | o’ | Oxygen in carbonyl (C=O) group |
| O | o | sp3 oxygen in ether or ester groups |
| O | o- | Oxygen in charged carboxylate (COO-) group |
| O | oh | Oxygen in hydroxyl (OH) group |
| O | o* | Oxygen in water molecule |
| O | op | Oxygen in aromatic rings. e.g. furan |
| O | of | Oxygen in |
| O | o$ | Oxygen atom for automatic parameter assignment |
| O | oc | sp3 oxygen in ether or acetals |
| O | oe | sp3 oxygen in ester |
| O | o3e | sp3 oxygen in three membered ring |
| O | o4e | sp3 oxygen in four membered ring |
| S | s | Sulfur in methionine (C-S-C) group |
| S | s1 | Sulfur involved in S-S disulfide bond |
| S | sh | Sulfur in sulfhydryl (-SH) group |
| S | sp | Sulfur in thiophene |
| S | s’ | Sulfur in thioketone (>C=S) group |
| S | s$ | Sulfur atom for automatic parameter assignment |
| S | sc | sp3 sulfur in methionines (C-S-C) group |
| S | s3e | Sulfur in three membered ring |
| S | s4e | Sulfur in four membered ring |
| S | s- | Sulfur bonded to something then bonded to another partial double O or S |
| P | p | General phosphorous atom |
| P | p$ | Phosphorous atom for automatic parameter assignment |
| Ca | ca+ | Calcium ion - Ca++, mass = mass of Ca - 2*electron mass. |
| F | f | Fluorine bonded to a carbon |
| Cl | cl | Chlorine bonded to a carbon |
| Br | br | Bromine bonded to a carbon |
| I | i | Covalently bound Iodine |
| Si | si | Silicon atom (General) |
| H | nu | NULL atom for relative free energy |
| Cl | Cl | Chloride ion Cl- |
| Br | Br | Bromide ion Br- |
| Na | Na | Sodium metal |
| Ar | ar | Argon |
| Si | sz | Silicon atom in zeolites |
| Si | sy | Tetrahedral Silicon atom in Clays |
| O | oz | Oxygen atom in zeolites |
| O | oy | Oxygen atom in Clays |
| Al | az | Tetrahedral Aluminum atom in zeolites |
| Al | ay | Octahedral Aluminum atom in Clays |
| Al | ayt | Tetrahedral Aluminum atom to be used with oy |
| P | pz | Phosphorous atom in zeolites |
| P | py | Phosphorous atom to be used with oy |
| Ga | ga | Gallium atom in zeolites |
| Ge | ge | Germanium atom in zeolites |
| Ti | tioc | Titanium (Octahedral) in zeolites |
| Ti | ti4c | Titanium (Octahedral) to be used with oy |
| Ti | titd | Titanium (Tetrahedral) in zeolites |
| Li | li+ | Lithium ion in zeolites |
| Li | lic+ | Lithium ion to be used with oy in Clays |
| Li | lioh | Lithium ion in water to be used with o* |
| Na | na+ | Sodium ion in zeolites |
| Na | nac+ | Sodium ion in Clays |
| Na | naoh | Sodium ion in water to be used with o* |
| K | k+ | Potassium ion in zeolites |
| K | koh | Potassium ion in water to be used with o* |
| Rb | rb+ | Rubidium ion in zeolites |
| Cs | cs+ | Cesium ion in zeolites |
| N | nh4+ | United atom type for ammonium ion to be used with oy |
| Mg | mg2+ | Magnesium ion in zeolites |
| Mg | mg2c | Octahedral Magnesium ion in Clays |
| Mn | mn4c | Manganese (IV) ion to be used with oy in Clays |
| Mn | mn3c | Manganese (III) ion to be used with oy in Clays |
| Co | co2c | Cobalt (II) ion to be used with oy in Clays |
| Ni | ni2c | Nickel (II) ion to be used with oy in Clays |
| Ca | ca2+ | Calcium ion in zeolites |
| Ca | ca2c | Calcium ion to be used with oy in Clays |
| Sr | sr2c | Strontium ion to be used with oy in Clays |
| Ba | ba2+ | Barium ion in zeolites |
| Cu | cu2+ | Copper(II) ion in zeolites |
| Fe | fe2c | Octahedral Fe(II) ion in clays |
| F | f- | Fluoride ion in zeolites |
| Be | beoh | Beryllium (II) in water to be used with o* |
| F | foh | Fluoride ion in water to be used with o* |
| Cl | cl- | Chloride ion in zeolites |
| Cl | cloh | Chloride ion in water to be used with o* |
| Cl | cly- | Chloride ion to be used with oy in Clays |
| Br | br- | Bromide ion in zeolites |
| I | i- | Iodide ion in zeolites |
| S | so4 | Sulfur in sulphate ion to be used with oz |
| S | so4y | Sulfur in sulphate ion to be used with oy in Clays |
| H | hocl | Hydrogen in hydroxyl group in Clays |
| Pd | pd2+ | Palladium(II) |
| V | vy | Tetrahedral Vanadium to be used with oy |
| Al | al | Aluminium metal |
| Na | Na | Sodium metal |
| Pt | Pt | Platinum metal |
| Pd | Pd | Palladium metal |
| Au | Au | Gold metal |
| Ag | Ag | Silver metal |
| Sn | Sn | Tin metal |
| K | K | Potassium metal |
| Li | Li | Lithium metal |
| Mo | Mo | Molybdenum metal |
| Fe | Fe | Iron metal |
| W | W | Tungsten metal |
| Ni | Ni | Nickel metal |
| Cr | Cr | Chromium metal |
| Cu | Cu | Copper metal |
| Pb | Pb | Lead metal |
2.19.3.2.4. Nacl.fr
This forcefield provides an illustration of the incorporation of a general inorganic forcefield description in the MedeA environment framework.
| Na | Na1+ | sodium atom |
| Cl | Cl1- | chlorine atom |
2.19.3.2.5. Clayff.frc clayff-dioctahedral.frc clayff-trioctahedral.frc
| H | h* | water hydrogen |
| H | ho | H hydroxyl hydrogen |
| O | o* | water oxygen |
| O | oh | hydroxyl oxygen |
| O | ob | Basal bridging oxygen |
| O | oa | Appical bridging oxygen |
| Si | st | Silicon in SiO2 |
| Al | ao | Aluminium in the octahedral sheet |
| Al | at | Aluminium in Zeolites |
| Mg | mgo | Magnesium in the octahedral sheet |
| Ca | cao | Calcium in the octahedral sheet |
| Fe | feo | iron in the octahedral sheet |
| Li | lio | Lithium in the octahedral sheet |
| O | obss | bridging oxygen with double substitution |
| O | obts | bridging oxygen with tetrahedral substitution |
| O | obos | bridging oxygen with octahedral substitution |
| O | ohs | hydroxyl oxygen with substitution |
| Ca | cah | hydroxide calcium |
| Mg | mgh | hydroxide magnesium |
| Na | Na | Sodium ion |
| K | K | Potassium ion |
| Cs | Cs | Cs+ ion |
| Ca | Ca | Ca2+ ion |
| Ba | Ba | Ba2+ ion |
| Cl | Cl | Cl- ion |
2.19.3.2.6. AlO_eam_coul.frc TaO_eam_coul.frc CeThUNpPuAmCmO_eam_coul.frc
These forcefields are known as the Streitz-Mintmire or charge-transfer ionic (CTIP) potentials [20] which combine EAM and Coulomb (charges described via Slater type orbitals instead of point charges) forcefields along with variable charge equilibration.
2.19.3.2.6.1. AlO_eam_coul.frc
| Al | Al | |
| O1 | O1 |
2.19.3.2.6.2. TaO_eam_coul.frc
| Ta | Ta | |
| O2 | O2 |
2.19.3.2.6.3. CeThUNpPuAmCmO_eam_coul.frc
| Ce | Ce | |
| Th | Th | |
| U | U | |
| Np | Np | |
| Pu | Pu | |
| Am | Am | |
| Cm | Cm | |
| O | O |
2.19.3.2.7. comb3.frc Si-O_JCP2016-comb3.frc
The 3rd generation charge-optimized many-body (COMB3) [21] forcefields are improvements over the previous generations of COMB forcefields. COMB3 contains an advanced bond order term for describing complex chemical reactions (bond breaking and formation), Coulomb with charge density described with Slater-type orbitals, and variable charge equilibration (atomic charges automatically assigned based on atomic surroundings).
2.19.3.2.7.1. comb3.frc
| Ti | Ti | Titanium |
| H | H | Hydrogen |
| C | C | Carbon |
| N | N | Nitrogen |
| O | O | Oxygen |
| Cu | Cu | Copper |
| Zn | Zn | Zinc |
| Zr | Zr | Zirconium |
| Si | Si | Silicon |
| Ti | Ti | Titanium |
| Al | Al | Aluminum |
| Ni | Ni | Nickel |
| Mo | Mo | Molybdenum |
| S | S | Sulfur |
| Pt | Pt | Platinum |
| Au | Au | Gold |
2.19.3.2.7.2. Si-O_JCP2016-comb3.frc
| O | O | Oxygen |
| Si | Si | Silicon |
2.19.3.3. Semiconductors
Forcefields for semiconductor materials. These forcefields don’t require bonds.
2.19.3.3.1. StillingerWeber.frc ZnCdTeSeHgS_Zhou_2013_StillingerWeber.frc
Stilllinger-Weber forcefields that allow for the simulation of various crystalline and amorphous solids. This forcefield uses an explicit angular term to assess nearest neighbor coordination (to include three-body forces) based on the local environment of simulated atoms [22].
2.19.3.3.1.1. StillingerWeber.frc
| Cd | Cd | cadmium |
| Ga | Ga | gallium |
| N | N | nitrogen |
| Si | Si | silicon |
| Te | Te | tellurium |
2.19.3.3.1.2. ZnCdTeSeHgS_Zhou_2013_StillingerWeber.frc
| Cd | Cd | cadmium |
| Zn | Zn | zinc |
| Te | Te | tellurium |
| Se | Se | selenium |
| Hg | Hg | mercury |
| S | S | sulfur |
2.19.3.3.2. Tersoff.frc SiO2-Si_Munetoh_2007_Tersoff.frc
Tersoff forcefields that allow for the simulation of various crystalline and amorphous solids. This forcefield uses a bond order term to assess nearest neighbor coordination (to include three-body forces) based on the local environment of simulated atoms. [23]
2.19.3.3.2.1. Tersoff.frc
| C | C | carbon |
| Ga | Ga | gallium |
| Ge | Ge | germanium |
| N | N | nitrogen |
| Si | Si | silicon, final parameters |
| Si | Si(B) | silicon, original parameters |
| Si | Si(C) | silicon, second set of parameters |
| O | O | oxygen atom |
2.19.3.3.2.2. SiO2-Si_Munetoh_2007_Tersoff.frc
| Si | Si | silicon, final parameters |
| O | O | oxygen atom |
2.19.3.3.2.3. REBO.frc
1st generation reactive bond order (REBO)[#30_24]_ forcefield closely related to Tersoff forcefields. It allows for simulations of Si with Cl, with Ar described via Moliere forcefield.
| Si | Si | silicon |
| Cl | Cl | chlorine |
| Ar | Ar | argon |
2.19.3.4. Metallic
The forcefields in this section don’t require bonds during atom type assignment and allow to study of metallic systems using the EAM (embedded atom model) description pioneered by Mike Baskes and others.
As noted above, the variability in local coordination inherent in inorganic systems (as opposed to organic systems) dictates that the creation of transferable forcefield descriptions is challenging for such systems. Hence, for each of the inorganic and metallic forcefield descriptions we recommend that the original references are consulted in order to assess the applicability of these descriptions to a particular system.
2.19.3.4.1. Zhou_2004.frc
This forcefield provides support for the following set of atoms and alloys composed of mixtures of these atoms. Zhou [25], with additions from Francis [26]
| Ag | Ag | silver |
| Al | Al | aluminum |
| Au | Au | gold |
| Co | Co | cobalt |
| Cu | Cu | copper |
| Fe | Fe | iron |
| Mg | Mg | magnesium |
| Mo | Mo | molybdenum |
| Ni | Ni | nickel |
| Pb | Pb | lead |
| Pd | Pd | palladium |
| Pt | Pt | platinum |
| Ta | Ta | tantalum |
| Ti | Ti | titanium |
| W | W | tungsten |
| Zr | Zr | zirconium |
2.19.3.4.2. EAM_Adams.frc
Li, Siegel, Adams, and Liu: [27]
| Al | Al | aluminum |
| Au | Au | gold |
| Cu | Cu | copper |
| Ni | Ni | nickel |
| Pd | Pd | palladium |
| Pt | Pt | platinum |
| Ta | Ta | tantalum |
2.19.3.4.5. md-eam.frc
Updated pair interaction function
| Zr | Zr | zirconium |
| Sn | Sn | tin |
| Cu | Cu | copper |
2.19.3.4.6. FeNiCr_Bonny_2011.frc
EAM forcefield for alloys containing Fe, Ni, and Cr [31]
| Fe | Fe | iron |
| Ni | Ni | nickel |
| Cr | Cr | cronium |
2.19.3.4.7. AlCo_Mishin_2013.frc AlNi_Mishin_2009.frc AlTi_Mishin_2003.frc AlCu_Cai_1996.frc AlMg_Adams_1997.frc
EAM forcefields (eam/alloy format) for alloys containing Al/Co [32], Al/Ni [33], Al/Ti [34], Al/Cu [35], and Al/Mg [36].
| Al | Al | aluminum |
| Co | Co | cobalt |
| Ni | Ni | nickel |
| Ti | Ti | titanium |
| Cu | Cu | copper |
| Mg | Mg | magnesium |
2.19.3.4.8. AlSiMgCuFe_MEAM.frc AuSi_MEAM.frc CH_MEAM.frc Cu_MEAM.frc FeC_MEAM.frc FeTiC_MEAM.frc Ni_MEAM.frc SiC_MEAM.frc W_MEAM.frc…… MEAM.frc
Modified EAM (MEAM) forcefields include an additional angular term for a more accurate description of metals and alloys, including Al/Si/Mg/Cu/Fe [37], Au/Si [38], C/H [39], Fe/C [40], Fe/Ti/C [41], W [42], and Si/C, Cu, and Ni from the LAMMPS website. A generic MEAM.frc is also included to be used with custom MEAM forcefield parameter sets.
2.19.3.4.8.1. AlSiMgCuFe_MEAM.frc
| Al | Al | aluminum |
| Si | Si | silicon |
| Mg | Mg | magnesium |
| Cu | Cu | copper |
| Fe | Fe | iron |
2.19.3.4.8.2. AuSi_MEAM.frc
| Au | Au | gold |
| Si | Si | silver |
2.19.3.4.8.3. CH_MEAM.frc
| C | C | carbon |
| H | H | hydrogen |
2.19.3.4.8.4. FeC_MEAM.frc
| Fe | Fe | iron |
| C | C | carbon |
2.19.3.4.8.5. FeTiC_MEAM.frc
| Fe | Fe | iron |
| Ti | Ti | titanium |
| C | C | carbon |
2.19.3.4.8.6. W_MEAM.frc
| W | W | tungstun |
2.19.3.4.8.7. SiC_MEAM.frc
| Si | Si | silicon |
| C | C | carbon |
2.19.3.4.8.8. Ni_MEAM.frc
| Ni | Ni | nickel |
2.19.3.4.8.9. Cu_MEAM.frc
| Cu | Cu | copper |
2.19.3.5. NIST Interatomic Potentials Repository
Detailed descriptions are shown in the file selection dialog. These files are distributed with consent from Chandler A. Becker, the maintainer of this repository [43], http://www.ctcms.nist.gov/potentials.
2.19.3.6. ReaxFF Forcefields
Reactive Forcefields (ReaxFF)[#30_44]_ is a family of well-established forcefields that simulate complex chemical reactions and charge transfer. It includes advanced bond terms over valence terms, shielded Coulomb, and variable charge equilibration.
2.19.3.6.1. AuOH.frc
ReaxFF forcefield for Au, AuOx and water [45] from LAMMPS potentials repository
| Au | Au | gold |
| O | O | oxygen |
| H | H | hydrogen |
2.19.3.6.2. CHO.frc
The well-established ReaxFF forcefield for combustion [46] simulations from LAMMPS potentials repository
| C | C | carbon |
| O | O | oxygen |
| H | H | hydrogen |
2.19.3.6.3. CHON.frc
The well-established ReaxFF forcefield for nitramines (RDX/HMX/TATB/PETN) [47] from LAMMPS potentials repository
| C | C | carbon |
| O | O | oxygen |
| H | H | hydrogen |
| N | N | nitrogen |
2.19.3.6.4. HONB.frc
ReaxFF forcefield for Ammonia Borane [48] from LAMMPS potentials repository
| B | B | boron |
| O | O | oxygen |
| H | H | hydrogen |
| N | N | nitrogen |
2.19.3.6.5. VCHO.frc
ReaxFF forcefield for V, VOx and water [49] from LAMMPS potentials repository
| V | V | vanadium |
| O | O | oxygen |
| H | H | hydrogen |
| C | C | carbon |
2.19.3.6.6. ZnOH.frc
ReaxFF forcefield for Zn, ZnOx and water [50] from LAMMPS potentials repository
| Zn | Zn | zinc |
| O | O | oxygen |
| H | H | hydrogen |
2.19.3.6.7. FeCOH.frc
ReaxFF forcefield for Fe, FeOx and water [51] from LAMMPS potentials repository
| V | V | vanadium |
| O | O | oxygen |
| H | H | hydrogen |
| C | C | carbon |
2.19.3.6.8. CHONSFPtClNi.frc
ReaxFF forcefield for fluorinated graphene, Pt [52] from LAMMPS potentials repository
| C | C | carbon |
| O | O | oxygen |
| H | H | hydrogen |
| N | N | nitrogen |
| S | S | sulfur |
| F | F | fluorine |
| Pt | Pt | platinum |
| Cl | Cl | chlorine |
| Ni | Ni | nickel |
2.19.4. The MaterialsDesign Forcefield Format - FRC
The advantages of the .frc format are as follows:
- automated atom type assignment using the templates section of the .frc file
- wildcards
- atom type equivalences for nonbonds, bonds, angles, torsions, etc.
- versioning: each parameter has its own version, so updates do not remove older parameters but override them
- includes: a user can modify a forcefield by including the original, adding parameters and, by using version numbers, override parameters in the original
The .frc format is much more compact and makes it easy to see and edit parameters. Wildcards are the ability to specify ‘*’ for an atom type. For example, the AUA forcefield specifies angles as C-CH2-C, where the terminal C can be almost any type of C atom, -CH3, -CH2-, -CH<, olefinic, ketone, etc. When you enumerate the permutations, it grows to be a very large list, which must be explicitly enumerated in e.g. GIBBS’ potparam.dat file.
With wildcards once specify one angle as *-CH2-*, where * matches any atom. More specific angles, like an alcohol *-C-O, including completely specific ones such as H-C-O take precedence in the obvious order. This also occurs in torsions, where typically the terminal atoms do not matter: *-CH2-CH2-*
For an example of the power of including forcefield files and version numbers, look at the oplsaa+.frc file, which includes the original oplsaa.frc, extensions published elsewhere (oplsaa-extended.frc), and adds some customs additions by MaterialsDesign:
Include FF
The first section is a definition of the OPLSAA+ forcefield, listing the functional forms and the sections of the file(s) that contain the parameters. In this case the forcefield uses the ‘OPLSAA’ section (which will come from oplsaa.frc via an include in oplsaa-extended.frc) and the ‘oplsaa+’ section (which is in this file). Next it includes the entire extended OPLS forcefield.
Atom Types
This section adds a new atom type for sp3 aliphatic carbon, ‘CT’. Each section of the file has the name of the section optionally followed by an increment to version number, 200 in this case. This increment is added to the version numbers in the section, so the practical version number of the ‘CT’ atom type is 1.0+200 = 201.0. Assuming that the OPLSS/AA forcefield uses version numbers less than 200, the ‘CT’ atom type would override any ‘CT’ atom types in OPLSS/AA. This allows you to take similar forcefields and let one override the other without modifying all the version numbers.
The next section defines equivalences, which simply say that when looking for the bond parameters for ‘CAh1’use ‘CA’, but when looking for bond increase parameters, use a different value for ‘CAh1’, Thus we have a new atom type that is much like sp2 aromatic carbon, but the bond increments are different.
Equivalences
Having a higher version overrides the previous line, and now we can use specific bond increment parameters for our ‘CAh1’ atom type.
Bond Increments
The last section of the example, the bond increment section, adds the bond parameters for our new ‘CAh1’ atom type. In addition, it adds or overrides some other bond parameters.
The final section concerns templates: It is by far the most complicated section, and unfortunately due to its nature cannot be versioned or added to. It must be taken as a whole unit because it specifies which atom type to assign to an atom in a structure, and hence is ‘aware’ of all the atom types in the forcefield and the relationship between them. Hence being monolithic.
If you need to modify the template section, copy the existing template section into the top level file and define this as the location of templates in the default section. Under normal circumstance you would inherit the template section from an included forcefield file and not touch it.
These extracts illustrate some of the important features. Each section defines how a local portion of the structure maps to an atom type. Each section is for an atom type and must contain the ‘template:’ line, which gives the topology.
Templates
As usual, ‘*’ is a wild card.
Parentheses around the template indicate that there may be other bonds to the atom that are not contemplated in the template; square brackets indicate that the template includes all bonds, and that extra bonds are not allowed. So the first template matches anything.
The ‘*’ wildcard matches any element and the surrounding parentheses allow any number of bonds.
The atom type is ‘?’ which is our shorthand for an atom for which there are no parameters. The next template is also quite simple: it matches any argon atom, regardless of whether it has bonds to it or not. If we wanted an explicit match for just argon atoms, i.e. without any bonds, we would surround the template with square brackets instead of a parenthesis.
Template for Ar
For bonds we use ‘-‘ for single bonds, ‘=’ for double bonds, ‘:’ for aromatic bonds, and ‘#’ for triple bonds; ‘~’ matches any bond order, i.e. it is a wildcard.
Template for C in esters/acids
Modifiers can narrow down the scope of wildcards: Allowed modifiers are hybridization, aromaticity, and elements:
Templates with wildcards
This template is quite specific for water. The square brackets both around the entire template and about the O and second H sees to that: there can be no other bonds anywhere.
Template with square brackets
This template is less specific, but fits e.g. CO 2and CS 2. It would also fit e.g. Ar-C-Ar and other nonsensical structures.
Template with square brackets and wildcards
This is a key issue in forcefields: they know what they do match, but not what they don’t!
With wildcards they tend to match many unintended things. So in the case of Ar-C-Ar, we would assign atom types just fine and (hopefully) still not be able to run because there would be missing Ar-C bond parameters and Ar-C-Ar angle parameters. On the other hand, if we had been lazy, and defined a set of generic bond parameters for ‘C-*’ and angle parameters for ‘*-C-*’ we would be off and running … garbage! It might be reasonable to have a catch-all angle term like ‘*-C-*’ since specific hybridization of the carbon atom (sp in this case) does roughly define the angle terms. But never a bond term like ‘C-*’! That is not reasonable since the bond length and strength depends on the second atom. And it is very dangerous, though the code will let you be foolish.
This brings us to more restraint use of wildcards: Here we see explicit tests that limit the power of the wildcards. The atom numbers are in the order the atoms appear in the templates, so the carbon of interest must be sp2; the two atoms other than oxygen that are bonded to it must be a C or H and an O or N. In other words this will match -C-C(=O)-OH, or H-C(=O)-OH, or -C-C(=O)-NH2 but not -C-C(=O)-C-. The modifiers for wildcards can be hybridization, which elements, and whether it is aromatic. At the moment, the code for recognizing hybridization and aromaticity is only partially complete.
Templates
The implementation in OPLS avoids the hybridization requirement and does not handle all cases, but goes through acids, esters, and, as shown amides:
Templates
Though not shown in this example, templates can match next nearest neighbors, etc. For example, the template for a carbon attached to an azide (-N3 group) looks like this:
Templates
The last section in the example is the precedence tree. An atom in a structure may match several templates, yielding different atom types. The precedence tree solves this ambiguity by providing a tree of atom types. The most specific match, i.e. the furthest from the trunk down a branch wins. The parentheses group the branches together but are admittedly rather hard to read.
Precedence tree
Other groups such as AMBER do have some level of typing engines, but mostly the bio-organic community relies on the regularity of peptides, proteins and DNA to use systematic atom naming schemes and ‘template libraries’ to match the atom types. Thus a protein is built from peptide fragments that already have the atom names and atom types assigned by hand. Since there are only twenty some amino acids, creating the fragment library is quite feasible. Proteins from the PDB also have systematic names for the atoms, so the template libraries match the atom types with the names. These are not, however, very general solutions.
| [1] | Huai Sun, Stephen J. Mumby, Jon R. Maple, and Arnold T. Hagler, “An Ab Initio CFF93 All-Atom Force Field for Polycarbonates,” Journal of the American Chemical Society 116, no. 7 (1994): 2978-2987. |
| [2] | William L Jorgensen, David S Maxwell, and Julian Tirado-Rives, “Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids,” Journal of the American Chemical Society 118, no. 45 (January 1996): 11225-11236. |
| [3] | MG Martin and IJ Siepmann, “Transferable Models for Phase Equilibria 1. United-Atom Description of N-Alkanes,” Journal of Physical Chemistry B 102 (1998): 2569. |
| [4] | Ganesh Kamath, Feng Cao, and Jeffrey J Potoff, “An Improved Force Field for the Prediction of the Vapor-Liquid Equilibria for Carboxylic Acids,” Journal of Physical Chemistry B 108, no. 37 (September 2004): 14130-14136. |
| [5] | John M Stubbs, Jeffrey J Potoff, and J Ilja Siepmann, “Transferable Potentials for Phase Equilibria. 6. United-Atom Description for Ethers, Glycols, Ketones, and Aldehydes,” Journal of Physical Chemistry B 108, no. 45 (November 2004): 17596-17605. |
| [6] | Collin D Wick, John M Stubbs, Neeraj Rai, and J Ilja Siepmann, “Transferable Potentials for Phase Equilibria. 7. Primary, Secondary, and Tertiary Amines, Nitroalkanes and Nitrobenzene, Nitriles, Amides, Pyridine, and Pyrimidine,” Journal of Physical Chemistry B 109, no. 40 (October 2005): 18974-18982. |
| [7] | Bin Chen, Jeffrey J Potoff, and J Ilja Siepmann, “Monte Carlo Calculations for Alcohols and Their Mixtures with Alkanes. Transferable Potentials for Phase Equilibria. 5. United-Atom Description of Primary, Secondary, and Tertiary Alcohols,” Journal of Physical Chemistry B 105, no. 15 (April 2001): 3093-3104. |
| [8] | Collin D Wick, Marcus G Martin, and J Ilja Siepmann, “Transferable Potentials for Phase Equilibria. 4. United-Atom Description of Linear and Branched Alkenes and Alkylbenzenes,” Journal of Physical Chemistry B 104, no. 33 (August 2000): 8008-8016. |
| [9] | MG Martin and IJ Siepmann, “Novel Configurational-Bias Monte Carlo Method for Branched Molecules. Transferable Potentials for Phase Equilibria. 2. United-Atoms Description of Branched Alkanes,” Journal of Physical Chemistry B 103 (1999): 4508. |
| [10] | N Lubna, G Kamath, J J Potoff, N Rai, and J I Siepmann, “Transferable Potentials for Phase Equilibria. 8. United-Atom Description for Thiols, Sulfides, Disulfides, and Thiophene,” Journal of Physical Chemistry B 109, no. 50 (2005): 24100-24107. |
| [11] | Katie A Maerzke, Nathan E Schultz, Richard B Ross, and J Ilja Siepmann, “TraPPE-UA Force Field for Acrylates and Monte Carlo Simulations for Their Mixtures with Alkanes and Alcohols,” Journal of Physical Chemistry B 113, no. 18 (May 7, 2009): 6415-6425. |
| [12] | H Sun, “COMPASS: an Ab Initio Force-Field Optimized for Condensed-Phase -Overview with Details on Alkane and Benzene Compounds,” Journal of Physical Chemistry B 102, no. 38 (September 1998): 7338-7364. |
| [13] | J\({\ddot{o}}\) rg-R diger Hill, Clive M Freeman, and Lalitha Subramanian, “Use of Force Fields in Materials Modeling,” in Reviews in Computational Chemistry, ed. by Kenny B Lipkowitz and Donald B Boyd, vol. 16, (Hoboken, NJ, USA: John Wiley & Sons, Inc., 2000), 141-216. |
| [14] | J R Maple, M J Hwang, T P Stockfisch, U Dinur, M Waldman, et al., “Derivation of Class II Force Fields. I. Methodology and Quantum Force Field for the Alkyl Functional Group and Alkane Molecules,” Journal of Computational Chemistry 15, no. 2 (February 1994): 162-182; M J Hwang, T P Stockfisch, and A T Hagler, “Derivation of Class II Force Fields. 2. Derivation and Characterization of a Class II Force Field, CFF93, for the Alkyl Functional Group and Alkane Molecules,” Journal of the American Chemical Society 116, no. 6 (1994): 2515-2525. |
| [15] | J R Maple, M J Hwang, T P Stockfisch, U Dinur, M Waldman, et al., “Derivation of Class II Force Fields. I. Methodology and Quantum Force Field for the Alkyl Functional Group and Alkane Molecules,” Journal of Computational Chemistry 15, no. 2 (February 1994): 162-182; M J Hwang, T P Stockfisch, and A T Hagler, “Derivation of Class II Force Fields. 2. Derivation and Characterization of a Class II Force Field, CFF93, for the Alkyl Functional Group and Alkane Molecules,” Journal of the American Chemical Society 116, no. 6 (1994): 2515-2525. |
| [16] | S.M. Woodley, P.D. Battle, J D Gale, and C Richard A Catlow, “The Prediction of Inorganic Crystal Structures Using a Genetic Algorithm and Energy Minimisation,” Physical Chemistry Chemical Physics 1, no. 10 (1999): 2535-2542. |
| [17] | Xin Xia, “Computational Modelling Study of Yttria-Stabilized Zirconia,” (University College London, 2010). |
| [18] | B W H van Beest, G J Kramer, and R A van Santen, “Force Fields for Silicas and Aluminophosphates Based on Ab Initio Calculations,” Physical Review Letters 64, no. 16 (April 1990): 1955-1958. |
| [19] | J\({\ddot{o}}\) rg-R diger Hill, Clive M Freeman, and Lalitha Subramanian, “Use of Force Fields in Materials Modeling,” in Reviews in Computational Chemistry, ed. by Kenny B Lipkowitz and Donald B Boyd, vol. 16, (Hoboken, NJ, USA: John Wiley & Sons, Inc., 2000), 141-216. |
| [20] | F. H. Streitz and J. W. Mintmire, “Electrostatic potentials for metal-oxide surfaces and interfaces” Phys. Rev. B 50, 11996 |
| [21] | T. Liang, T.-R. Shan, Y.-T. Cheng, B. D. Devine, M. Noordhoek, Y. Li, Z. Lu, S. R. Phillpot, and S. B. Sinnott, Mat. Sci. & Eng: R 74, 255-279 (2013). |
| [22] | Frank H Stillinger and Thomas A Weber, “Computer Simulation of Local Order in Condensed Phases of Silicon,” Physical Review B 31, no. 8 (1985): 5262-5271; A. B\({\grave{o}}\) r\({\grave{o}}\) and A. Serra, “On the Atomic Structures, Mobility and Interactions of Extended Defects in GaN: Dislocations, Tilt and Twin Boundaries,” Philosophical Magazine 86, no. 15 (2006): 2159-2192. |
| [23] | J Tersoff, “New Empirical Approach for the Structure and Energy of Covalent Systems,” Physical Review B 37, no. 12 (1988): 6991-7000; J Tersoff, “Empirical Interatomic Potential for Silicon with Improved Elastic Properties,” Physical Review B 38, no. 14 (1988): 9902-9905; J Tersoff, “Modeling Solid-State Chemistry: Interatomic Potentials for Multicomponent Systems,” Physical Review B 39, no. 8 (1989): 5566-5568; J Tersoff, “Erratum: Modeling Solid-State Chemistry: Interatomic Potentials for Multicomponent Systems,” Physical Review B 41, no. 5 (1990): 3248-3248; “Modelling of Compound Semiconductors: Analytical Bond-Order Potential for Gallium, Nitrogen and Gallium Nitride,” Journal of Physics: Condensed Matter 15, no. 32 (2003): 5649. |
| [24] | D. W. Brenner Phys. Rev. B 42, 9458 (1990); D. Humbird and D. B. Graves, J. Chem. Phys. 120, 2405 (2004) |
| [25] | X Zhou, R Johnson, and H Wadley, “Misfit-Energy-Increasing Dislocations in Vapor-Deposited CoFe/NiFe Multilayers,” Physical Review B 69, no. 14 (April 2004). |
| [26] | M F Francis, M N Neurock, X W Zhou, J J Quan, H N G Wadley, et al., “Atomic Assembly of Cu/Ta Multilayers: Surface Roughness, Grain Structure, Misfit Dislocations, and Amorphization,” Journal of Applied Physics 104, no. 3 (2008): 034310. |
| [27] | Youhong Li, Donald J Siegel, James Adams, and Xiang-Yang Liu, “Embedded-Atom-Method Tantalum Potential Developed by the Force-Matching Method,” Physical Review B 67, no. 12 (2003). |
| [28] | Yuri Mishin and Diana Farkas, “Atomistic Simulation of Point Defects and Diffusion in B2 NiAl Part1: Point Defect Energetics,” Philosophical Magazine A 75, no. 1 (1997): 169-185; Yuri Mishin and Diana Farkas, “Atomistic Simulation of Point Defects and Diffusion in B2 NiAl Part2: Diffusion Mechanisms,” Philosophical Magazine A 75, no. 1 (1997): 187-199. |
| [29] | G J Ackland, G Tichy, V Vitek, and M W Finnis, “Simple N-Body Potentials for the Noble Metals and Nickel,” Philosophical Magazine A 56, no. 6 (December 1987): 735-756. |
| [30] | M I Mendelev and G J Ackland, “Development of an Interatomic Potential for the Simulation of Phase Transformations in Zirconium,” Philosophical Magazine A 87, no. 5 (May 2007): 349-359. |
| [31] | G. Bonny, D. Terentyev, R.C. Pasianot, S. Ponc\({\grave{o}}\) , and A. Bakaev, “Interatomic potential to study plasticity in stainless steels: the FeNiCr model alloy.” Modelling and simulation in materials science and engineering, 19, 085008 (2011) |
| [32] | Purja Pun, G. P., Yamakov, V., and Mishin, Y. (2015). Interatomic potential for the ternary Ni-Al-Co system and application to atomistic modeling of the B2-L1 0 martensitic transformation. Modelling Simul. Mater. Sci. Eng., 23(6), 065006 |
| [33] | G.P. Purja Pun and Y. Mishin, “Development of an interatomic potential for the Ni-Al system,” Phil. Mag. 89, 3245 (2009). |
| [34] | R.R. Zope and Y. Mishin, “Interatomic potentials for atomistic simulations of the Ti-Al system,” Phys. Rev. B 68, 024102 (2003) |
| [35] | X.-Y. Liu, C.-L. Liu, and L.J. Borucki, “A new investigation of copper’s role in enhancing Al-Cu interconnect electromigration resistance from an atomistic view,” Acta Mat. 47, 3227-3231 (1999) |
| [36] | X.-Y. Liu, P.P. Ohotnicky, J.B. Adams, C. Lane Rohrer, R.W. Hyland, Jr., “Anisotropic surface segregation in Al-Mg alloys,” Surf. Sci. 373, 357-370 (1997) |
| [37] | B. Jelinek, S. Groh, M. Horstemeyer, J. Houze, S.G. Kim, G.J. Wagner, A. Moitra, and M.I. Baskes, “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Phys. Rev. B 85, 245102 (2012) |
| [38] | J. Godet, C. Furgeaud, L. Pizzagalli, M. Demkowicz, “Uniform tensile elongation in Au-Si core-shell nanowires”, Extreme Mechanics Letters (2016) |
| [39] | S. Nouranian, M.A. Tschopp, S.R. Gwaltney, M.I. Baskes, and M.F. Horstemeyer, “An interatomic potential for saturated hydrocarbons based on the modified embedded-atom method,” Physical Chemistry Chemical Physics 16, 6233 (2014). |
| [40] | L.S.I. Liyanage, S.-G. Kim, J. Houze, S. Kim, M.A. Tschopp, M.I. Baskes, and M.F. Horstemeyer, “Structural, elastic, and thermal properties of cementite (Fe13C) calculated using a modified embedded atom method,” Phys. Rev. B 89, 094102 (2014) |
| [41] | Kim, H.-K., Jung, W.-S., and Lee, B.-J. (2009). Modified embedded-atom method interatomic potentials for the Fe-Ti-C and Fe-Ti-N ternary systems. Acta Materialia, 57(11), 3140-3147. |
| [42] | Lee, Baskes, Kim, Cho. Phys. Rev. B, 64, 184102 (2001) |
| [43] | Chandler A Becker, Francesca Tavazza, Zachary T Trautt, and Robert A Buarque de Macedo, “Considerations for Choosing and Using Force Fields and Interatomic Potentials in Materials Science and Engineering,” Current Opinion in Solid State and Materials Science 17 (December 2013): 277-283. |
| [44] | A.C.T. van Duin, S. Dasgupta, F. Lorant, and W. A. Goddard, ReaxFF: A reactive force field for hydrocarbons, Journal of Physical Chemistry A 105, 9396-9409 (2001); Chenoweth, A.C.T. van Duin, and W.A. Goddard, ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation, Journal of Physical Chemistry A 112, 1040-1053 (2008) |
| [45] | Keith, J. A. et al. Phys Rev B 2010, 81, 235404 |
| [46] | Chenoweth, A.C.T. van Duin, and W.A. Goddard, ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation, Journal of Physical Chemistry A 112, 1040-1053 (2008) |
| [47] | Strachan et al, Phys Rev Lett, 91, 098301 (2003) |
| [48] | Weismiller, van Duin, Lee, Yetter, J Phys Chem A, 114, 5485-5492 (2010) |
| [49] | Chenoweth et al, J Phys Chem C, 112, 14645-14654 (2008) |
| [50] | Raymand, van Duin, Spangberg, Goddard and Hermansson, Surf Sci, 604, 741-752 (2010) |
| [51] | Aryanpour, van Duin and Kubicki, J Phys Chem A, 114, 6298-6307 (2010) |
| [52] | Singh, Phys Rev AB 87, 104114 (2013) |
| download: | pdf |
|---|